")
Back to Journals » Journal of Pain Research » Volume 8
New insights into the use of currently available non-steroidal anti-inflammatory drugs
Authors Brune K, Patrignani P
Received 30 September 2014
Accepted for publication 27 November 2014
Published 20 February 2015 Volume 2015:8 Pages 105—118
DOI https://doi.org/10.2147/JPR.S75160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Michael Schatman
Kay Brune,1 Paola Patrignani2
1Department of Experimental and Clinical Pharmacology and Toxicology, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany; 2Department of Neuroscience, Imaging and Clinical Sciences, Center of Excellence on Aging, G d’Annunzio University, Chieti, Italy
Abstract: Non-steroidal anti-inflammatory drugs (NSAIDs), which act via inhibition of the cyclooxygenase (COX) isozymes, were discovered more than 100 years ago. They remain a key component of the pharmacological management of acute and chronic pain. The COX-1 and COX-2 isozymes have different biological functions; analgesic activity is primarily (although not exclusively) associated with inhibition of COX-2, while different side effects result from the inhibition of COX-1 and COX-2. All available NSAIDs, including acetaminophen and aspirin, are associated with potential side effects, particularly gastrointestinal and cardiovascular effects, related to their relative selectivity for COX-1 and COX-2. Since all NSAIDs exert their therapeutic activity through inhibition of the COX isozymes, strategies are needed to reduce the risks associated with NSAIDs while achieving sufficient pain relief. A better understanding of the inhibitory activity and COX-1/COX-2 selectivity of an NSAID at therapeutic doses, based on pharmacokinetic and pharmacodynamic properties (eg, inhibitory dose, absorption, plasma versus tissue distribution, and elimination), and the impact on drug tolerability and safety can guide the selection of appropriate NSAIDs for pain management. For example, many NSAIDs with moderate to high selectivity for COX-2 versus COX-1 can be administered at doses that maximize efficacy (~80% inhibition of COX-2) while minimizing COX-1 inhibition and associated side effects, such as gastrointestinal toxicity. Acidic NSAIDs with favorable tissue distribution and short plasma half-lives can additionally be dosed to provide near-constant analgesia while minimizing plasma concentrations to permit recovery of COX-mediated prostaglandin production in the vascular wall and other organs. Each patient's clinical background, including gastrointestinal and cardiovascular risk factors, should be taken into account when selecting appropriate NSAIDs. New methods are emerging to assist clinicians in the selection of appropriate NSAIDs and their doses/schedules, such as biomarkers that may predict the response to NSAID treatment in individual patients.
Keywords: cyclooxygenase inhibitors, cyclooxygenase selectivity, diclofenac, pharmacodynamics, pharmacokinetics, pain therapy
Introduction
Pain represents a major public health problem worldwide,1–3 with chronic pain affecting approximately 27% of the adult population in Europe4 and more than 100 million adults in the United States.1 Undertreated acute pain can be associated with an increased risk of deleterious health consequences (eg, delayed wound healing, immune dysfunction, cardiovascular problems related to the stress response, and respiratory problems, such as pneumonia) and the development of chronic pain.5 In addition, unrelieved chronic severe pain can negatively impact an individual’s quality of life, day-to-day functioning, sleep quality, interpersonal relationships, and work productivity, and is associated with a substantial economic burden.1,6
There are two major options for the pharmacological management of pain: non-steroidal anti-inflammatory drugs (NSAIDs), which act via inhibition of cyclooxygenase (COX) isozymes, and opiate/opioid analgesics.7,8 NSAIDs, which are among the most widely used medications worldwide,9,10 are often preferred because of their low abuse potential, robust efficacy, and long history of clinical use.11 Guidelines for pain management tend to be specific to medical conditions or settings. Current guidelines for chronic low back pain and osteoarthritis generally recommend the use of oral acetaminophen (or topical NSAIDs for osteoarthritis) for first-line pain management, with other oral NSAIDs recommended in the first-line or second-line setting depending on the specific guideline; some guidelines also recommend opioids, although typically not as first-line therapy.8,12–15 It has, however, recently been shown that acetaminophen is not measurably effective in acute low back pain (ie, not more effective than placebo).16 Other work in patients with osteoarthritis demonstrated that ibuprofen was associated with greater improvements compared with acetaminophen in measures of osteoarthritis pain, function, and quality of life.17 Acetaminophen and other oral NSAIDs are also recommended for acute pain, such as postoperative pain management.7
The first non-opioid analgesics (phenazone, paracetamol, and aspirin) were found by serendipity more than 100 years ago.18 Later, systematic research identified their mode of action as inhibition of prostanoid production.18 The development of animal models of pain and inflammation led to the discovery of a host of new NSAIDs (eg, diclofenac, ibuprofen, ketoprofen, naproxen), many of which were acidic, highly protein-bound compounds that generally showed similar therapeutic effects and side effects related to their inhibitory activity on prostanoid synthesis.18
NSAIDs inhibit prostanoid biosynthesis through their activity on the COX enzymes COX-1 and COX-2.19 The two COX isoforms (COX-1 and COX-2) have different functions, and the inhibition of these isoforms results in different therapeutic effects and side effects.11 Efforts to avoid the gastrointestinal side effects associated with COX-1 inhibition11 led to the development of selective COX-2 inhibitors called “coxibs”, which were designed to inhibit COX-2 while sparing COX-1 at therapeutic doses.18 Evidence of increased risk of myocardial infarction and other thrombotic events led to concerns over the cardiovascular safety of coxibs, resulting in the withdrawal of rofecoxib from the market in 2004.10 However, traditional NSAIDs, which inhibit COX-2 without sustained inhibition of platelet COX-1, may also be associated with an increased risk of cardiovascular events, particularly at high doses.11,20–23
Although novel compounds are being developed (eg, COX-inhibiting nitrous oxide donors) to improve safety and tolerability, it is unlikely that an ideal oral NSAID that avoids all potential side effects will be available in the near future.11,24 Thus, strategies are needed to reduce the risks associated with currently available NSAIDs. Drug choice should be driven by the clinical and demographic features (eg, age) of the patient, and the lowest effective dose of drug should be used for the shortest period of time25 to reduce the risk of cardiovascular and gastrointestinal side effects related, at least in part, to drug exposure.11
The development of whole-blood assays of COX isozyme inhibition in vitro has proved useful for differentiating NSAIDs based on their pharmacodynamic features, such as COX-2 versus COX-1 selectivity (ie, experimental COX isoenzyme selectivity),26–28 while ex vivo assessment after NSAID dosing allows evaluation of COX isozyme selectivity achieved at circulating drug levels. Additionally, the use of biochemical markers of COX isozyme inhibition in vivo, such as the assessment of major enzymatic urinary metabolites of the parent prostanoid, permits assessment of the actual inhibition of prostanoid biosynthesis following NSAID dosing. The use of these biomarkers of COX inhibition have provided a mechanistic interpretation of the efficacy, adverse events, and interindividual variability associated with NSAID treatment.21,22,29–31
This review provides an overview of these and other key aspects that differentiate orally administered NSAIDs, with a focus on the role of these factors in establishing the safety and tolerability of individual NSAIDs for analgesia. Characterization of the pharmacokinetic and pharmacodynamic differences between these compounds and their relationship with the administered oral dose may allow for better tailoring of NSAID therapy to individual patient needs.
Mechanism of action of NSAIDs: COX-1 and COX-2 inhibition
The anti-inflammatory, analgesic, and antipyretic activities of NSAIDs are mediated by their inhibition of prostanoid biosynthesis.11,32,33 Prostanoids are synthesized from arachidonic acid, a fatty acid present in cell membranes as a phospholipid ester.11,19,32 COX isozymes convert arachidonic acid first to prostaglandin (PG)G2 and then to PGH2, which undergoes a series of subsequent conversion reactions, ultimately producing five bioactive prostanoids, ie, PGD2, PGE2, PGF2α, PGI2 (prostacyclin), and thromboxane A2 (TxA2).19 These bioactive prostanoids exhibit various cell-specific and tissue-specific activities through their interaction with different receptors, mediating a range of diverse, and often opposing, physiological and pathological processes, including induction and resolution of the inflammatory response, protection of and damage to the gastrointestinal mucosa, promotion and inhibition of blood clotting and atherosclerosis, and renal control of blood pressure and renal disease.23,34
As described previously, there are two distinct isoforms of COX: the constitutively expressed COX-1 isoform and the inducible COX-2 isoform.11,32,35 COX-1 is present in the majority of cells and tissues, including the endothelium, monocytes, gastrointestinal epithelial cells, and platelets.32 In contrast, COX-2 is constitutively expressed in only a few tissues.32 However, expression of COX-2 is upregulated in a variety of cells and tissues, such as vascular endothelium, rheumatoid synovial endothelial cells, monocytes, and macrophages, during inflammation through the actions of various inflammatory mediators (eg, bacterial endotoxins, tumor necrosis factor α, interleukins); the increase in COX-2 protein levels is the primary driving force for enhanced production of prostanoids at inflammatory sites.32,35 Although COX-2 is the primary pathway, evidence suggests that COX-1 may also contribute to the initial phase of prostanoid-dependent pain and inflammation; in vivo, COX-1 is expressed, along with COX-2, in circulating inflammatory cells and in inflamed tissue.19,34 The roles of COX-1 and COX-2 in different systems are summarized in Figure 1.
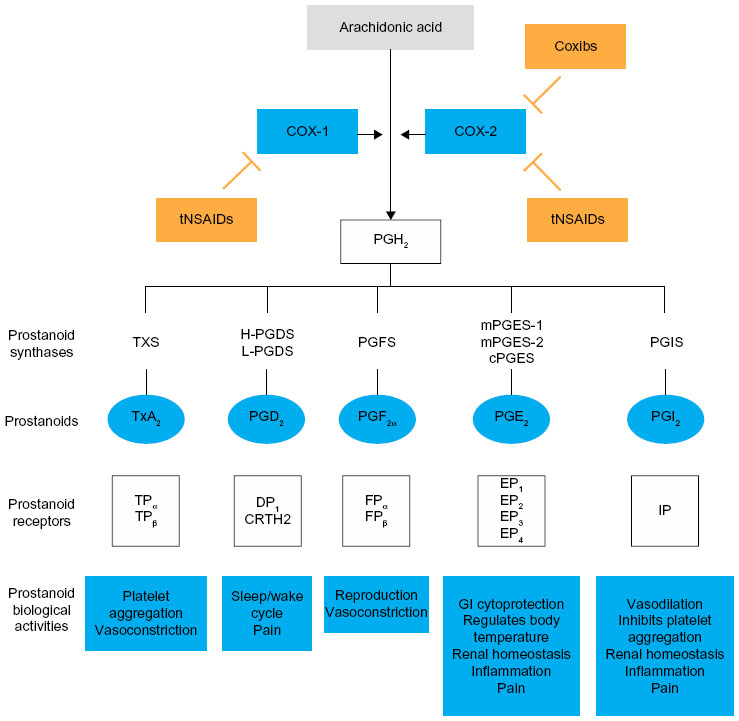 | Figure 1 Mechanism of action of NSAIDs. |
During the inflammatory response, COX-dependent prostanoids play a role in the development of hyperalgesia. PGE2 and PGI2 increase the sensitivity of pain receptors (or nociceptors) in the periphery and enhance the activity of various pain mediators.34,36 Peripheral inflammation is also associated with upregulation of COX-2 and an increase in PGE2 in the central nervous system, while contributing to the development of central hyperalgesia.34 The role of COX-2 in the development of central hyperalgesia has been demonstrated in an engineered mouse model, in which conditional deletion of the gene for COX-2 in neurons and glial cells of the central nervous system resulted in the reduction of peripheral inflammation–induced COX-2 expression in the spinal cord and of mechanical hypersensitivity related to joint movement or tenderness to touch.37
In general, NSAIDs inhibit prostanoid synthesis by competitive and transient inhibition of arachidonic acid binding to the COX active site.19 The therapeutic effects of NSAIDs largely result from COX-2 inhibition at sites of inflammation, while many of the side effects associated with NSAIDs, particularly gastrointestinal side effects, are attributed to inhibition of the protective effects of prostanoids produced by COX-1.11,23,33 Although all NSAIDs generally act by inhibition of arachidonic acid binding at COX, the mechanism of action of aspirin (acetylsalicylic acid) differs fundamentally from that of non-aspirin NSAIDs in that it causes an irreversible inactivation of COX-1 and COX-2 in most patients, preventing the oxidative conversion of arachidonic acid to PGG2 and PGH2.38 In non-nucleated platelets, this effect confers persistent inhibition of COX-1–mediated TxA2 production and platelet function throughout the dosing interval.38,39 Acetaminophen also differs from other NSAIDs in that it has no measurable anti-inflammatory effects at therapeutic doses,40 but primarily inhibits prostanoid-mediated hyperalgesia through suppression of PGE2 biosynthesis.40–42
Inhibition of prostanoid biosynthesis by NSAIDs is responsible for the therapeutic activities of these drugs;11,32 however, this inhibitory activity also suppresses other effects of these prostanoids, resulting in the side effects associated with NSAID treatment.22,23,33 For example, inhibition of PGE2 production may reduce hyperalgesia but may also reduce the tissue-protective effects of this prostanoid in various organs, including the gastrointestinal tract, vascular wall, kidney, and lung.11
Pharmacokinetics and pharmacodynamics of NSAIDs
COX isozyme selectivity
Different NSAIDs display different levels of selectivity for the COX-1 and COX-2 isoforms in vitro as a consequence of the chemical features of the drug.11,21,22,33 Selectivity for the COX-1 and COX-2 isoforms, as well as the extent of inhibition reached after dosing with NSAIDs, can be determined using whole-blood assays.27,28,43 Maximal platelet COX-1 activity is assessed via measurement of TxB2 (the inactive metabolite of TxA2, generated non-enzymatically) formed in whole blood allowed to clot for 1 hour at 37°C. This assay uses endogenously generated thrombin to release arachidonic acid from platelets; arachidonic acid is converted to PGH2 by COX-1 and then transformed to TxA2 by the activity of thromboxane synthase.27,28 To evaluate maximal monocyte COX-2 activity, PGE2 levels are measured in whole blood following 24 hours of stimulation with exogenously added lipopolysaccharide to induce COX-2 expression in circulating monocytes.27
Several studies have been performed to define the relationships between the extent of inhibition of COX-1 and COX-2 in whole blood after dosing with NSAIDs (COX isozyme inhibition ex vivo) and the degree of inhibition of prostanoid biosynthesis in vivo by assessing urinary levels of the major enzymatic metabolite of TxB2, 11-dehydro-TxB2, and the major metabolite of prostacyclin, 2,3-dino-6-keto-PGF1α.44,45 The use of low-dose aspirin, which is a selective inhibitor of platelet COX-1, and coxibs has shown that 11-dehydro-TxB2 is primarily of platelet origin (~70%), while 2,3-dino-6-keto-PGF1α is primarily derived by vascular COX-2 (~60%). Importantly, inhibition of 11-dehydro-TxB2 after dosing with aspirin is initially detected when the maximal capacity of platelet COX-1 is reduced by >97%.45 Thus, the relationship between inhibition of platelet TxB2 biosynthesis ex vivo and in vivo is non-linear (Figure 2A).44 In contrast, the relationship between inhibition of COX-2 activity measured ex vivo and the reduction in PGI2 biosynthesis measured in vivo is apparently linear (Figure 2B).19,45
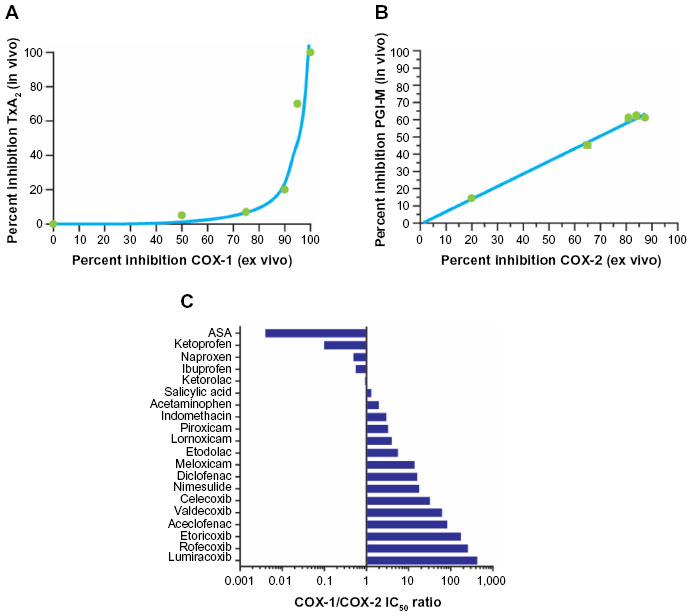 | Figure 2 Selectivity of NSAIDs for COX-1 and COX-2. |
This knowledge allows prediction of the clinical consequences of COX isozyme inhibition by NSAIDs. NSAIDs may affect platelet function in vivo when the inhibition of platelet COX-1 ex vivo is >97%.45 This effect occurs primarily with low-dose aspirin and persists throughout the dose interval. Naproxen, which is a reversible inhibitor of COX isozymes and has a higher potency for COX-1 than COX-2 and a long half-life (~14 hours), may have an aspirin-like effect on platelet function in some individuals when administered at high (500 mg twice daily) doses.46 The use of whole-blood assays allows for characterization of COX isozyme selectivity of NSAIDs in vitro. Concentration-dependent relationships for inhibition of whole-blood COX-1 and COX-2 activities as a function of the addition of an NSAID in vitro allow for assessment of half-maximal inhibitory concentration (IC50) values for COX-1 and COX-2, as well as calculation of IC50 ratios for COX-1/COX-2. COX-2 selectivity assessed in human whole blood in vitro is a continuous variable, and it is not possible to separate traditional NSAIDs from coxibs (Figure 2C).21,27,42,47–52
All NSAIDs are essentially COX-2 inhibitors with differing degrees of COX-1 inhibition as a “side effect”. From a clinical perspective, COX-2 selectivity can be considered as a variable describing the probability of sparing COX-1 activity and avoiding associated side effects (eg, in the gastrointestinal mucosa and platelets) at therapeutic concentrations of the NSAID.53 Drugs that inhibit COX-1 and COX-2 with comparable potency (known as nonselective NSAIDs; eg, ibuprofen and ketoprofen) will not spare COX-1 activity after dosing, while drugs with intermediate COX-2 selectivity (eg, nimesulide, meloxicam, diclofenac, celecoxib) or highly selective COX-2 inhibitors (eg, rofecoxib, etoricoxib, lumiracoxib [not available in the United States]) have greater potential for sparing COX-1 activity.23
Although in vitro COX-2 selectivity is a chemical feature of the NSAID, COX-2 selectivity measured in whole blood following dosing depends on the dose administered.23,54 NSAIDs are administered at doses that are not bioequivalent with respect to the extent of inhibition of monocyte COX-2 (which represents a target for drug efficacy) obtained at maximal concentration of drug in the systemic circulation after dosing (Cmax). In particular, NSAIDs with short half-lives (eg, diclofenac) are often given at higher doses to extend their duration of clinical efficacy (analgesic or anti-inflammatory effect). As shown in Figure 3, diclofenac 50 mg three times daily is associated with a higher inhibitory effect (>90% inhibition) over 8 hours following dosing compared with other NSAIDs.54 Different NSAIDs are associated with a differential impact of platelet COX-1 (which represents an index of drug-associated side effects) depending upon their COX isozyme selectivity. Thus, administration of high-dose diclofenac and meloxicam, which have comparable and intermediate COX-2 selectivity in vitro, results in partial inhibition of COX-1. Further, the anti-inflammatory and full analgesic effects of NSAIDs are associated with plasma concentrations that inhibit whole-blood COX-2 activity by 80% (IC80).55 Therefore, for drugs such as diclofenac, lowering the dose will lead to circulating concentrations near the COX-2 IC80 values, which correlate with adequate analgesia while reducing COX-1–mediated side effects.23,53
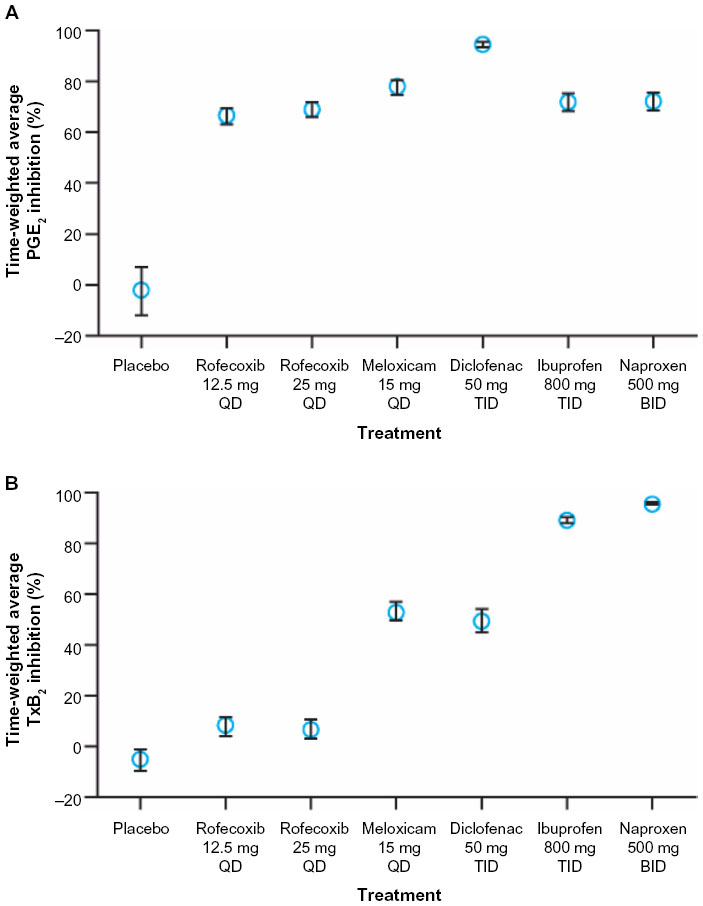 | Figure 3 Average inhibition of whole-blood COX-2 activity (A) and COX-1 activity (B) by NSAIDs. Inhibition was observed over 8 hours following dosing with different NSAIDs. |
Absorption, distribution, and elimination
In addition to dose, the therapeutic activity and side effects of NSAIDs depend on their absorption, distribution, and elimination; these pharmacokinetic parameters can differ substantially between different NSAIDs.56 As with any analgesic class, the rate of absorption is a key factor in the selection of an NSAID; those with rapid absorption are preferable for most patients, particularly those with severe or acute pain.56 For example, celecoxib, which has a relatively slow rate of absorption, can be administered at standard doses to effectively treat osteoarthritis pain, but is not ideal for treatment of acute pain as it takes considerable time for absorption and often requires a loading dose to achieve clinically meaningful analgesia.56
The distribution of NSAIDs in injured tissues, blood, and other areas of the body is a particularly crucial consideration for ensuring activity at the site of inflammation, as well as reducing the risk of side effects unrelated to therapeutic activity on COX enzymes throughout the body.56,57 NSAIDs can be classified as acidic or non-acidic based on their chemical structure, and the acidity of the drug can have an effect on its distribution. NSAIDs with acidic functional groups (eg, diclofenac, ibuprofen, ketoprofen) and with a high degree of protein binding have been shown to selectively accumulate and persist at sites of inflammation,56–59 while non-acidic NSAIDs (eg, acetaminophen, celecoxib, rofecoxib) tend to be distributed homogenously throughout the body.56 Acidic NSAIDs with a high degree of protein binding may remain in inflamed tissues or synovial fluid for a longer time than in the plasma (Figure 4). For example, diclofenac 75 mg (enteric-coated) reaches peak concentrations in the synovial fluid that exceed plasma concentrations at 4 hours after oral administration in patients with arthritis;60 these high and persistent synovial levels are maintained above or near the expected IC80 for COX-2 for up to 12 hours after dosing, with rapid disappearance of diclofenac from the plasma, even after repeated dosing.60 The persistence of certain NSAIDs (eg, diclofenac, ibuprofen) in the synovial fluid is associated with a sustained therapeutic effect despite relatively rapid clearance from the plasma, vascular wall, and kidneys, indicating that accumulation at sites of inflammation may allow for continued anti-inflammatory and analgesic activity.57 The localization of COX activity to inflamed tissue and the resulting ability to use lower doses than might otherwise be required also minimizes COX inhibition at sites associated with potential side effects (eg, hepatic, renal, cardiovascular, and gastrointestinal adverse effects).57,58
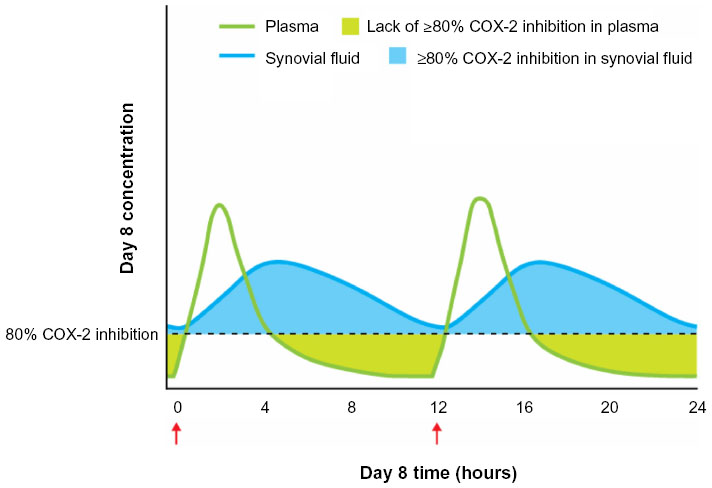 | Figure 4 NSAID concentration in inflamed tissue/synovial fluid and plasma. |
The accumulation of NSAIDs at sites of inflammation and accompanying therapeutic effects are also affected by their pharmacokinetic properties (including extended-release or immediate-release formulations). Acidic NSAIDs with short plasma half-lives (eg, diclofenac, ibuprofen, ketoprofen) may be associated with tolerability benefits compared with drugs with a long plasma half-life due to rapid clearance from the plasma and non-targeted tissues, allowing for recovery of COX activity in other tissues (eg, production of vasoprotective prostanoids by endothelial COX-2) even while the drug continues to actively inhibit COX-2 at sites of inflammation. The use of excessively high doses or slow-release formulations of these analgesics could negate these benefits.21,29,56,58 Further studies using in vivo markers of COX-2 inhibition (such as urinary metabolites of PGE2 and PGI2) should ideally be conducted to verify this hypothesis. Persistence in the plasma is also a key factor with respect to the gastrointestinal safety and tolerability of NSAIDs.61,62
Interactions of NSAIDs with other drugs
NSAIDs are also differentiated by their potential for pharmacodynamic interactions with other drugs, such as aspirin, corticosteroids, and selective serotonin reuptake inhibitors, which are often administered concomitantly for different reasons.56,63,64 Aspirin irreversibly acetylates the COX active site, preventing generation of TxA2, a prothrombotic prostanoid involved in platelet activation and aggregation.38,39 NSAIDs characterized by a high affinity for platelet COX-1 (eg, ibuprofen, naproxen) may interfere with the antiplatelet effects of aspirin; in contrast, this effect has not been detected with NSAIDs having intermediate or high selectivity for COX-2.65–67 Moreover, no interaction has been detected with low-dose paracetamol.66 Although further randomized controlled trials are needed to confirm the clinical relevance of these pharmacodynamic interactions, an analysis of the medical records of 7,107 patients with cardiovascular disease who received aspirin alone or aspirin plus an NSAID indicated that the risks of all-cause mortality and cardiovascular-related mortality were significantly higher for patients who received aspirin plus ibuprofen compared with those who received aspirin alone (P>0.05), but not for patients who received aspirin plus diclofenac or aspirin plus any other NSAID (drugs not specified).68
NSAID therapy in patients taking anticoagulant therapy with coumarins (vitamin K antagonists) should also be approached with caution. Coadministration of NSAIDs with coumarins may be associated with an increased risk of bleeding complications, particularly for naproxen.69 The increased risk of bleeding associated with concomitant use of NSAIDs and coumarins (eg, warfarin) results from displacement of the anticoagulant from plasma proteins by the NSAID, thereby increasing the plasma concentration of free coumarin and associated anticoagulant activity and bleeding risk.70
NSAID safety and tolerability
Gastrointestinal safety and tolerability
Gastrointestinal complications are among the most common and well-known side effects of NSAID treatment and generally result from inhibition of COX-1.11,32 COX-1 is expressed throughout the gastrointestinal tract and mediates the production of mucosal PGs that confer a number of protective effects, including increasing blood flow to the mucosa and promoting mucus and bicarbonate secretion;11,32 inhibition of COX-1 may therefore result in an increased susceptibility for mucosal damage (eg, bleeding).11,23,32 Additionally, COX-2 may promote healing of gastric lesions; thus, inhibition of COX-2 may also play a role in the formation of ulcers.23
A recent meta-analysis of data for more than 220,000 patients from 280 placebo-controlled trials of NSAIDs showed that all evaluated NSAIDs (coxibs, diclofenac, ibuprofen, and naproxen) increased the early risk of upper gastrointestinal complications (eg, ulcer perforations, bleeding, obstructions) to some extent.20 There is, however, considerable variability in the risk of gastrointestinal complications between NSAIDs.29,71,72 Results of a 2012 meta-analysis of 28 observational studies showed a low risk (relative risk [RR] >2) of upper gastrointestinal complications for aceclofenac, celecoxib, and ibuprofen; intermediate risk (RR 2–4) for diclofenac, meloxicam, and ketoprofen, among others; high risk (RR 4–5) for tenoxicam, naproxen, indomethacin, and diflunisal; and the highest risk (RR >5) for piroxicam, ketorolac, and azapropazone.71 In keeping with results for individual NSAIDs, drugs that have greater selectivity for COX-2 than COX-1 generally have been associated with a lower risk of upper gastrointestinal complications than other NSAIDs.20,29,73 In the recent meta-analysis of data from more than 220,000 patients, the predicted absolute annual risk of upper gastrointestinal complications was lower for NSAIDs with greater COX-2 selectivity (coxibs and diclofenac) compared with non-selective NSAIDs (Figure 5).20
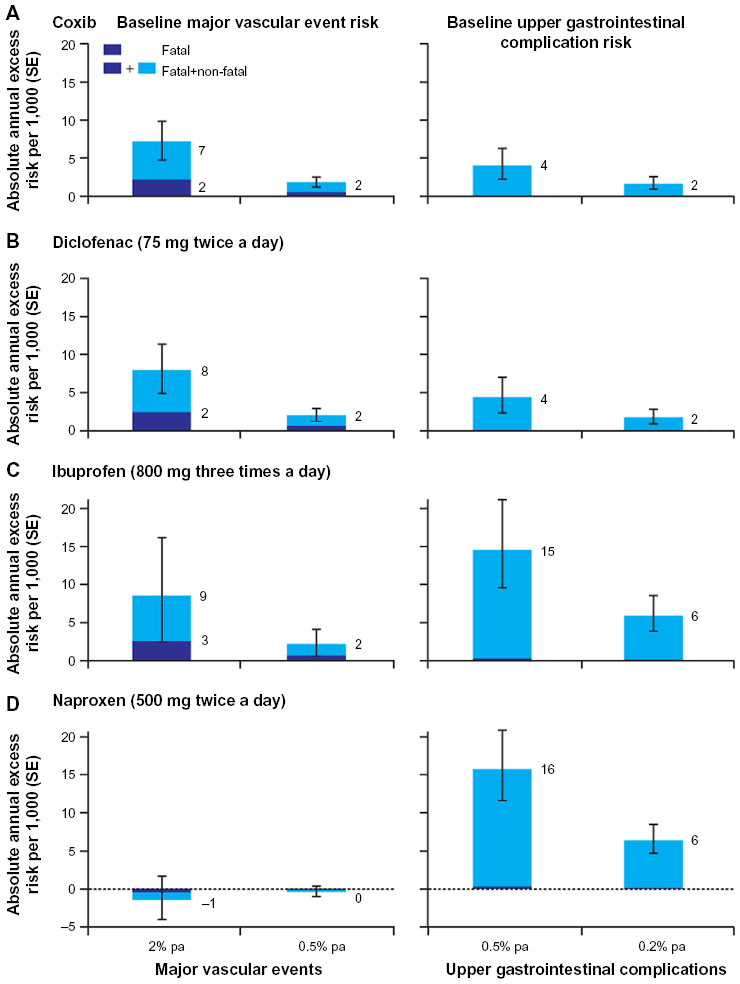 | Figure 5 Predicted annual absolute risks of major vascular events or upper gastrointestinal complications with long-term, high-dose therapy. |
Drugs with a long plasma half-life and/or slow-release formulations have been associated with an increased risk of upper gastrointestinal bleeding, likely due to persistent exposure of the gastrointestinal tract to circulating NSAIDs.29 In addition, the use of high daily doses of NSAIDs is also associated with a two- to three-fold increase in the risk of upper gastrointestinal complications relative to low daily doses,71 possibly due to dose-related effects on the levels of COX-1 and COX-2 inhibition.54,74 Because both COX-1 and COX-2 are associated with the production of cytoprotective prostanoids, simultaneous and sustained inhibition of both COX isozymes by NSAIDs may translate into a profound suppression of these prostanoids and promote damage to the gastrointestinal tract.23,29
Cardiovascular safety and tolerability
Although aspirin confers cardioprotective effects, most other NSAIDs are associated with an increased risk of cardiovascular adverse events, including hypertension, myocardial infarction, stroke, and heart failure.10,11,75–77 In the meta-analysis of data from more than 220,000 patients, a significant increase in the risk of major vascular events was observed for high doses of coxibs and diclofenac, and was largely related to increased risk of major coronary events; in contrast, neither ibuprofen or naproxen were associated with an increased risk of major vascular events, although ibuprofen was associated with an increase in major coronary events comparable with that of coxibs and diclofenac (Figure 5).20 The cardiovascular safety findings for diclofenac may reflect the use of doses above the IC80 for COX-2 (typically 150 mg/day).20 At lower doses, there is little evidence for an increased risk of cardiovascular events with diclofenac.78 A recent meta-analysis of cardiovascular risk and total daily diclofenac dose from eleven studies demonstrated a linear relationship between diclofenac dose and risk of experiencing a cardiovascular event, with only a small (although significant) increase in risk observed with low doses of diclofenac compared with not taking an NSAID.79
Like the risk of gastrointestinal complications, the risk of cardiovascular complications can be affected by drug exposure.21,77 In a 2008 observational study, the risk of myocardial infarction was increased with higher doses and with exposure to slow-release formulations of NSAIDs, even after adjusting for the dose.21 Based on these and other safety findings, the American Heart Association recommends patients take the lowest effective dose of NSAIDs for the shortest duration of time.25
The cardiovascular risk conferred by non-aspirin NSAIDs is likely associated with inhibition of COX-2–mediated production of PGI2, which has a cardioprotective role in the circulatory system, promoting vasodilation, preventing platelet activation and cell adhesion,22,23 and counteracting the action of TxA2, as well as several other stimuli.22 The antiplatelet effects of aspirin are mediated by irreversible inhibition of COX-1, resulting in profound and persistent reduction of TxA2 production and TxA2-dependent platelet activation (for several days after aspirin administration).38,66 However, for most other traditional NSAIDs and coxibs, COX-1 inhibition is only transient and insufficient to translate into inhibition of atherothrombosis.23 The exception is naproxen, which has a long half-life and potently inhibits platelet COX-1 activity sufficiently to prevent platelet aggregation at high doses;44,80 however, unlike aspirin, naproxen is a reversible inhibitor of COX-1 with variable effects across dosing intervals.80 Moreover, naproxen profoundly affects systemic PGI2 biosynthesis, while aspirin (at a low dosage) has only a marginal effect on biosynthesis of PGI2.80 Thus, as shown in the clinical data, naproxen is neutral with regard to cardiovascular risk, while aspirin is associated with a protective role.19
Biomarkers to predict drug responses
The use of biochemical markers of COX inhibition may assist clinicians in the rational selection of an appropriate NSAID dose to achieve efficacy; this dose should be the lowest effective dose to limit the risk of adverse events.25 However, differences in the efficacy and tolerability of NSAIDs have been observed between individuals,21,29,30,47 resulting from genetic factors that affect the pharmacokinetics and pharmacodynamics of NSAIDs (Figure 6).26 For example, cytochrome (CYP) enzymes are responsible for the metabolism of approximately 70%–80% of all drugs, including several NSAIDs,81,82 and interindividual differences in CYP450 expression can therefore result in altered metabolism of some NSAIDs.26 A number of single-nucleotide polymorphisms in the genetic sequence of CYP2C9 have been described,83 and the product of one of these altered genes, CYP2C9*2, is associated with reduced metabolism of celecoxib and an accompanying increase in CYP2C9*2 plasma concentration within 4 hours of administration in healthy individuals.30
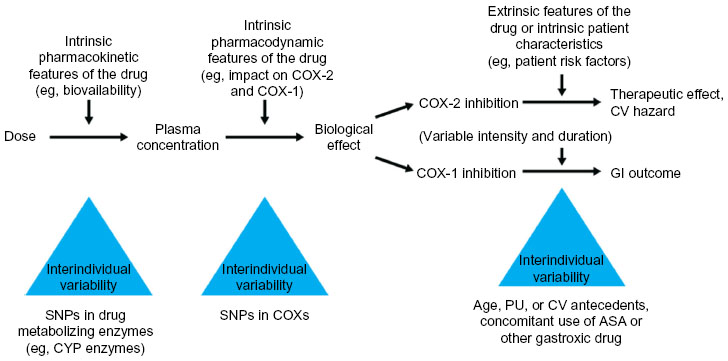 | Figure 6 Determinants and sources of variability in the individual response to an NSAID. |
In addition to interindividual variability in COX inhibition, the individual’s clinical background, particularly gastrointestinal and cardiovascular risk factors, should also be considered as important predictors of the risk of adverse reactions due to NSAIDs. Together, these factors should be used to select an NSAID and dose that will be both effective and well tolerated,26,55 and to help guide individualized treatment strategies.
Studies are ongoing to verify the feasibility of using of biochemical markers of COX isozyme inhibition ex vivo and in vivo together with genetic biomarkers to identify individuals at accelerated risk of developing cardiovascular and gastrointestinal adverse events by NSAID administration.
Conclusion
NSAIDs are among the most commonly prescribed and used medications worldwide.9,10 However, they are associated with safety and tolerability concerns.23 There are a number of factors that need to be considered in the selection of an NSAID, including its potency, selectivity for the COX-1 and COX-2 isoforms, pharmacokinetic properties, pharmacodynamic interactions, and the overall impact of these features on tolerability and safety. In addition to the gastrointestinal and cardiovascular safety of NSAIDs, the negative effects of inhibiting prostanoid production on other body systems should be considered. Emerging factors, including biomarkers, may assist clinicians in the selection of appropriate doses and types of NSAIDs. Finally, the individual’s clinical background, particularly gastrointestinal and cardiovascular risk factors, should be considered as important determinants that may exacerbate adverse reactions to NSAIDs. The risk factors for cardiovascular and gastrointestinal side effects of NSAIDs are summarized in Table 1. NSAIDs should be used with caution in patients with risk factors for atherothrombotic events. The long-term use of higher doses of NSAIDs represents an independent risk factor for cardiovascular and gastrointestinal adverse events;25,84 therefore, the lowest dose of an NSAID should be used, particularly in patients with other known risk factors.25
 | Table 1 Risk factors for NSAID gastrointestinal and cardiovascular side effects |
Current knowledge of factors affecting the safety and tolerability of NSAIDs, including the impact of selectivity, dose, and pharmacokinetics, is being used to guide the development of novel formulations of NSAIDs that address some of these tolerability concerns. For example, a novel immediate-release diclofenac formulation, which has a short half-life, acidic profile, and COX-2 selectivity, has been developed that allows for the use of lower doses (thus reducing systemic exposure and the potential for adverse events) together with rapid attainment of Cmax, providing sustained analgesia with a rapid onset.85 Strategies to mitigate the risks and maximize the therapeutic benefits associated with NSAIDs should continue to be employed, such as use of the lowest effective dose for the shortest period of time, use of immediate-release formulations, avoidance of known drug interactions (eg, concomitant use with corticosteroids, low-dose aspirin, or other antiplatelet/anticoagulation events), and limited use of NSAIDs with high gastrointestinal toxicity.23,86,87 Additional strategies for minimizing the risks associated with NSAIDs in individuals with gastrointestinal and/or cardiovascular risk factors are summarized in Table 2.
 | Table 2 Prevention strategies in patients with cardiovascular and/or gastrointestinal risk factors treated with NSAIDs |
Author contributions
KB and PP developed the concepts for the article, provided evaluation of relevant literature information, revised the manuscript critically for intellectual content, and approved the final version of the manuscript for publication.
Disclosures
Editorial support for the writing of this manuscript was provided by Kimberly Brooks, PhD, of SciFluent, and was funded by Iroko Pharmaceuticals, LLC. The authors retained full editorial control over the content of the manuscript. KB has provided consulting services to and received honoraria from Iroko Pharmaceuticals. PP has received grants from Associazione Italiana per la Ricerca sul Cancro, Ministero dell’Istruzione, dell’Università e della Ricerca; has received speaker fees from Bayer and Iroko Pharmaceuticals; and has provided consulting services to and received honoraria from Iroko Pharmaceuticals.
References
Committee on Advancing Pain Research, Care, and Education; Institute of Medicine. Relieving Pain in America. Washington, DC, USA: National Academies Press; 2011. | |
Reid KJ, Harker J, Bala MM, et al. Epidemiology of chronic non-cancer pain in Europe: narrative review of prevalence, pain treatments and pain impact. Curr Med Res Opin. 2011;27(2):449–462. | |
Tsang A, Von Korff M, Lee S, et al. Common chronic pain conditions in developed and developing countries: gender and age differences and comorbidity with depression-anxiety disorders. J Pain. 2008;9(10):883–891. | |
Leadley RM, Armstrong N, Lee YC, Allen A, Kleijnen J. Chronic diseases in the European Union: the prevalence and health cost implications of chronic pain. J Pain Palliat Care Pharmacother. 2012;26(4):310–325. | |
Ead H. Improving pain management for critically ill and injured patients. Dynamics. 2005;16(3):26–31. | |
McCarberg BH, Nicholson BD, Todd KH, Palmer T, Penles L. The impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet survey. Am J Ther. 2008;15(4):312–320. | |
American Society of Anesthesiologists Task Force on Acute Pain Management. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2012;116(2):248–273. | |
American Society of Anesthesiologists Task Force on Chronic Pain Management; American Society of Regional Anesthesia and Pain Medicine. Practice guidelines for chronic pain management: an updated report by the American Society of Anesthesiologists Task Force on Chronic Pain Management and the American Society of Regional Anesthesia and Pain Medicine. Anesthesiology. 2010;112(4):810–833. | |
Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10(4):287–333. | |
Conaghan PG. A turbulent decade for NSAIDs: update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol Int. 2012;32(6):1491–1502. | |
Atchison JW, Herndon CM, Rusie E. NSAIDs for musculoskeletal pain management: current perspectives and novel strategies to improve safety. J Manag Care Pharm. 2013;19(9 Suppl A):S3–S19. | |
Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478–491. | |
Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64(4):465–474. | |
National Institute for Health and Care Excellence. Clinical guideline 88. Low back pain: early management of persistent non-specific low back pain. 2009. Available from: http://www.nice.org.uk/guidance/cg88. Accessed November 29, 2014. | |
National Institute for Health and Care Excellence. Clinical guideline 177. Osteoarthritis: care and management in adults. 2014. Available from: http://www.nice.org.uk/guidance/cg59. Accessed Novemeber 29, 2014. | |
Williams CM, Maher CG, Latimer J, et al. Efficacy of paracetamol for acute low-back pain: a double-blind, randomised controlled trial. Lancet. 2014;384(9954):1586–1596. | |
Doherty M, Hawkey C, Goulder M, Gibb I, Hill N, Aspley S, Reader S. A randomised controlled trial of ibuprofen, paracetamol or a combination tablet of ibuprofen/paracetamol in community-derived people with knee pain. Ann Rheum Dis. 2011;70(9):1534–1541. | |
Brune K, Hinz B. The discovery and development of antiinflammatory drugs. Arthritis Rheum. 2004;50(8):2391–2399. | |
Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. | |
Bhala N, Emberson J, Merhi A, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769–779. | |
García Rodríguez LA, Tacconelli S, Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J Am Coll Cardiol. 2008;52(20):1628–1636. | |
Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116(1):4–15. | |
Patrignani P, Tacconelli S, Bruno A, Sostres C, Lanas A. Managing the adverse effects of nonsteroidal anti-inflammatory drugs. Expert Rev Clin Pharmacol. 2011;4(5):605–621. | |
Korotkova M, Jakobsson PJ. Characterization of microsomal prostaglandin E synthase 1 inhibitors. Basic Clin Pharmacol Toxicol. 2014;114(1):64–69. | |
Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634–1642. | |
Bruno A, Tacconelli S, Patrignani P. Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic Clin Pharmacol Toxicol. 2014;114(1):56–63. | |
Patrignani P, Panara MR, Greco A, et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther. 1994;271(3):1705–1712. | |
Patrono C, Ciabattoni G, Pinca E, et al. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb Res. 1980;17(3–4):317–327. | |
Masso Gonzalez EL, Patrignani P, Tacconelli S, García Rodríguez LA. Variability among nonsteroidal antiinflammatory drugs in risk of upper gastrointestinal bleeding. Arthritis Rheum. 2010;62(6):1592–1601. | |
Fries S, Grosser T, Price TS, et al. Marked interindividual variability in the response to selective inhibitors of cyclooxygenase-2. Gastroenterology. 2006;130(1):55–64. | |
Patrono C, Patrignani P, García Rodríguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001;108(1):7–13. | |
Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs):cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s. | |
Fitzgerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345(6):433–442. | |
Smyth EM, Grosser T, Wang M, Yu Y, Fitzgerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50 Suppl:S423–S428. | |
Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56(3):387–437. | |
Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388(6643):678–682. | |
Vardeh D, Wang D, Costigan M, et al. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119(2):287–294. | |
Meek IL, van de Laar MA, Vonkeman HE. Non-steroidal anti-inflammatory drugs: an overview of cardiovascular risks. Pharmaceuticals. 2010;3:2146–2162. | |
Patrono C, Baigent C, Hirsh J, Roth G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest. 2008;133(6 Suppl):199S–233S. | |
Hinz B, Cheremina O, Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J. 2008;22(2):383–390. | |
Anderson BJ. Paracetamol (acetaminophen):mechanisms of action. Paediatr Anaesth. 2008;18(10):915–921. | |
Sciulli MG, Seta F, Tacconelli S, et al. Effects of acetaminophen on constitutive and inducible prostanoid biosynthesis in human blood cells. Br J Pharmacol. 2003;138(4):634–641. | |
Blain H, Boileau C, Lapicque F, et al. Limitation of the in vitro whole blood assay for predicting the COX selectivity of NSAIDs in clinical use. Br J Clin Pharmacol. 2002;53(3):255–265. | |
Reilly IA, Fitzgerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69(1):180–186. | |
Capone ML, Tacconelli S, Sciulli MG, et al. Human pharmacology of naproxen sodium. J Pharmacol Exp Ther. 2007;322(2):453–460. | |
Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302–1308. | |
Panara MR, Renda G, Sciulli MG, et al. Dose-dependent inhibition of platelet cyclooxygenase-1 and monocyte cyclooxygenase-2 by meloxicam in healthy subjects. J Pharmacol Exp Ther. 1999;290(1):276–280. | |
Dovizio M, Bruno A, Tacconelli S, Patrignani P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013; 191:39–65. | |
Panara MR, Padovano R, Sciulli MG, et al. Effects of nimesulide on constitutive and inducible prostanoid biosynthesis in human beings. Clin Pharmacol Ther. 1998;63(6):672–681. | |
Patrignani P, Panara MR, Sciulli MG, Santini G, Renda G, Patrono C. Differential inhibition of human prostaglandin endoperoxide synthase-1 and -2 by nonsteroidal anti-inflammatory drugs. J Physiol Pharmacol. 1997;48(4):623–631. | |
Tacconelli S, Capone ML, Sciulli MG, Ricciotti E, Patrignani P. The biochemical selectivity of novel COX-2 inhibitors in whole blood assays of COX-isozyme activity. Curr Med Res Opin. 2002;18(8):503–511. | |
Patrignani P, Campestrini J, Branson J, et al. Lumiracoxib is a selective inhibitor of cyclooxygenase-2 in patients with osteoarthritis. Ann Rheum Dis. 2004;63:1419–1426. | |
Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82(1–4):85–94. | |
Van Hecken A, Schwartz JI, Depre M, et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J Clin Pharmacol. 2000;40(10):1109–1120. | |
Huntjens DR, Danhof M, la Pasqua OE. Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology (Oxford). 2005;44(7):846–859. | |
Brune K, Renner B, Hinz B. Using pharmacokinetic principles to optimize pain therapy. Nat Rev Rheumatol. 2010;6(10):589–598. | |
Brune K, Furst DE. Combining enzyme specificity and tissue selectivity of cyclooxygenase inhibitors: towards better tolerability? Rheumatology (Oxford). 2007;46(6):911–919. | |
Brune K. Persistence of NSAIDs at effect sites and rapid disappearance from side-effect compartments contributes to tolerability. Curr Med Res Opin. 2007;23(12):2985–2995. | |
Rolf C, Engstrom B, Beauchard C, Jacobs LD, Le Liboux A. Intra-articular absorption and distribution of ketoprofen after topical plaster application and oral intake in 100 patients undergoing knee arthroscopy. Rheumatology (Oxford). 1999;38(6):564–567. | |
Benson MD, Aldo-Benson M, Brandt KD. Synovial fluid concentrations of diclofenac in patients with rheumatoid arthritis or osteoarthritis. Semin Arthritis Rheum. 1985;15(2 Suppl 1):65–67. | |
Goldstein JL, Eisen GM, Lewis B, Gralnek IM, Zlotnick S, Fort JG. Video capsule endoscopy to prospectively assess small bowel injury with celecoxib, naproxen plus omeprazole, and placebo. Clin Gastroenterol Hepatol. 2005;3(2):133–141. | |
Hawkey C, Burnett I, Gold MS, et al. Endoscopic evaluation of the gastro-duodenal tolerance of short-term analgesic treatment with 25 mg diclofenac-K liquid capsules. Aliment Pharmacol Ther. 2012;35(7):819–827. | |
Mort JR, Aparasu RR, Baer RK. Interaction between selective serotonin reuptake inhibitors and nonsteroidal antiinflammatory drugs: review of the literature. Pharmacotherapy. 2006;26(9):1307–1313. | |
Verbeeck RK. Pharmacokinetic drug interactions with nonsteroidal anti-inflammatory drugs. Clin Pharmacokinet. 1990;19(1):44–66. | |
Anzellotti P, Capone ML, Jeyam A, et al. Low-dose naproxen interferes with the antiplatelet effects of aspirin in healthy subjects: recommendations to minimize the functional consequences. Arthritis Rheum. 2011;63(3):850–859. | |
Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345(25):1809–1817. | |
Renda G, Tacconelli S, Capone ML, et al. Celecoxib, ibuprofen, and the antiplatelet effect of aspirin in patients with osteoarthritis and ischemic heart disease. Clin Pharmacol Ther. 2006;80(3):264–274. | |
MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361(9357):573–574. | |
Penning-van Beest F, Erkens J, Petersen KU, Koelz HR, Herings R. Main comedications associated with major bleeding during anticoagulant therapy with coumarins. Eur J Clin Pharmacol. 2005;61(5–6):439–444. | |
Devi AM, Varghese AK, Sriram S, et al. Evaluation of in vitro interactions of warfarin and duloxetine with selected coadministered nsaids in bovine serum albumin. Int J Pharm Pharm Sci. 2011;3(1):197–199. | |
Castellsague J, Riera-Guardia N, Calingaert B, et al. Individual NSAIDs and upper gastrointestinal complications: a systematic review and meta-analysis of observational studies (the SOS project). Drug Saf. 2012;35(12):1127–1146. | |
García Rodríguez LA, Jick H. Risk of upper gastrointestinal bleeding and perforation associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343(8900):769–772. | |
García Rodríguez LA, Barreales TL. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology. 2007;132(2):498–506. | |
Hinz B, Brune K. Can drug removals involving cyclooxygenase-2 inhibitors be avoided? A plea for human pharmacology. Trends Pharmacol Sci. 2008;29(8):391–397. | |
Yu Y, Ricciotti E, Grosser T, Fitzgerald GA. The translational therapeutics of prostaglandin inhibition in atherothrombosis. J Thromb Haemost. 2009;7 Suppl 1:222–226. | |
McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296(13):1633–1644. | |
McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8(9):e1001098. | |
Brune K. Diclofenac: increase of myocardial infarctions at low doses? Pharmacoepidemiol Drug Saf. 2014;23(3):326–328. | |
Odom DM, Mladsi DM, Saag KG, et al. Relationship between diclofenac dose and risk of gastrointestinal and cardiovascular events: meta-regression based on two systematic literature reviews. Clin Ther. 2014;36(6):906–917. | |
Capone ML, Tacconelli S, Sciulli MG, et al. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation. 2004;109(12):1468–1471. | |
Guzey C, Spigset O. Genotyping as a tool to predict adverse drug reactions. Curr Top Med Chem. 2004;4(13):1411–1421. | |
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–141. | |
The Human Cytochrome P450 (CYP) Allele Nomenclature Committee. The human cytochrome P450 (CYP) allele nomenclature database. 2014. Available from: http://www.cypalleles.ki.se/. Accessed November 29, 2014. | |
Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340(24):1888–1899. | |
Gibofsky A, Silberstein S, Argoff C, Daniels S, Jensen S, Young CL. Lower-dose diclofenac submicron particle capsules provide early and sustained acute patient pain relief in a phase 3 study. Postgrad Med. 2013;125(5):130–138. | |
American College of Rheumatology. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Arthritis Rheum. 2000;43(9):1905–1915. | |
Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation. 2008;118(18):1894–1909. | |
Justice E, Carruthers DM. Cardiovascular risk and COX-2 inhibition in rheumatological practice. J Hum Hypertens. 2005;19(1):1–5. | |
Dixon DA, Blanco FF, Bruno A, Patrignani P. Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 2013;191:7–37. | |
Marnett LJ. The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol. 2009;49:265–290. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.