")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
Neu Perspectives, Therapies, and Challenges for Metastatic HER2-Positive Breast Cancer
Authors Salkeni MA, Rizvi W , Hein K, Higa GM
Received 24 June 2021
Accepted for publication 20 August 2021
Published 24 September 2021 Volume 2021:13 Pages 539—557
DOI https://doi.org/10.2147/BCTT.S288344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Mohamad Adham Salkeni,1 Wajeeha Rizvi,2 Kyaw Hein,3 Gerald M Higa4
1Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA; 2Department of Internal Medicine, West Virginia University, Morgantown, WV, USA; 3Department of Business, Lamar University, Houston, TX, USA; 4Departments of Clinical Pharmacy and Medicine, West Virginia University, Morgantown, WV, USA
Correspondence: Gerald M Higa
Departments of Clinical Pharmacy and Medicine, West Virginia University, Morgantown, WV, 26506, USA
Email [email protected]
Abstract: Even though gene amplification or protein overexpression occurs in approximately one-fifth of all breast cancers, the discovery of HER2 has, nevertheless, had profound implications for the disease. Indeed, the characterization of the receptor resulted in a number of significant advances. Structurally, unique features provided avenues for the development of numerous compounds with target-specificity; molecularly, biological constructs revealed a highly complex, internal signal transduction pathway with regulatory effects on tumor proliferation, survival, and perhaps, even resistance; and clinically, disease outcomes manifested its predictive and prognostic value. Yet despite the receptor’s utility, the beneficial effects are diminished by tumor recurrence after neo- or adjuvant therapy as well as losses resulting from the inability to cure patients with metastatic disease. What these observations suggest is that while tumor response may be partially linked to uncoupling cell surface message reception and nuclear gene expression, as well as recruitment of the innate immune system, disease progression and/or resistance may involve a reprogrammable signaling mainframe that elicits alternative growth and survival signals. This review attempts to meld current perceptions related to HER2-positive metastatic breast cancer with particular attention to current biological insights and therapeutic challenges.
Keywords: ado-trastuzumab emtansine, ErbB, Fam-trastuzumab deruxtecan, HER2, lapatinib, margetuximab, neratinib, neu, pertuzumab, PHESGO, trastuzumab, tucatinib
Perspectives
Neu-Setting
Although four decades old, the neu story is inherently correct in phraseology. Linked to a neuroblastoma cell line established in a mouse model,1 the neu-encoded receptor was found to be markedly similar to the epidermal growth factor receptor, the protein-product of v-ErbB.2,3 Not only was the connection between both genes reinforced by the significant homology of the receptors, but human EGFR-related was also the basis for the derivation of the moniker for HER family members.4 More importantly, HER2 is not only considered one of the two most important pharmacologic targets in breast cancer but like the estrogen receptor (ER), HER2 has predictive and prognostic values as well. And even if the receptor is a unique molecular signature, the other HER-family members may induce surreptitiously virulent effects on tumor tissue, the relevance of which will be discussed later.
Early on, the authors clarify nomenclature related to genes and proteins mentioned above. Biologically, ErbB2 refers to the gene, which encodes HER2 in humans and Neu in rodents. Hence, while ErbB2 applies to both species, the terms HER2 and Neu distinguish the species. Nonetheless, the use of the term “Neu” as a primary descriptor in this paper shows reverence to mice in advancing human understanding of the gene’s role in tumorigenesis.
The primary aim of this article targets the ongoing conundrum related to the genetic, molecular, and clinical challenges associated with HER2-positive metastatic breast cancer (HER2+ mBC). Whereas a discussion of the receptor provides the requisite insight, supplemental details were incorporated to describe the manner in which other elements could influence HER2-mediated signaling. And because of the historically abject clinical outcomes, an inclusive discourse of therapies approved or in clinical trials was also integrated into the paper. In essence, this review was constructed in a manner to provide the reader with the opportunity to learn a little about the current status regarding tumor biology and more about how molecular pharmacologic studies improve our understanding of the disease, and appreciate why complete eradication of the tumor continues to be elusive. The last section focuses on the issue related to the development of resistance and brain metastasis, two biological perturbations that continue to evade scientific resolution. Accordingly, the authors accessed and critically evaluated many creditable manuscripts in order to compose this paper.
Neu-Mapping
Positioned on chromosome 17, precisely at q21.2, HER2 harbors an encryption for a 1255 amino acid, 185 kD transmembrane receptor tyrosine kinase (p185HER2 RTK).5 Even though molecularly distinct, all HER family members have three structural zones including a short transmembrane portion, which bridges the external ligand-binding and internal catalytic kinase domains (Figure 1).6 Contrary to the notion that ligand binding, which promotes receptor coupling, is the initiating stimulus for downstream signal activation, it is now accepted that spatial arrangement or receptor configuration is the determining factor for catalytic phosphorylation.7,8 Even so, the ultimate cellular response depends on complex processing of signals derived from homo- or hetero-dimerized HER2 as well as bi-directional crosstalk with other receptors that may be mechanistically involved in survival, growth, and resistance of this subtype of breast cancer.
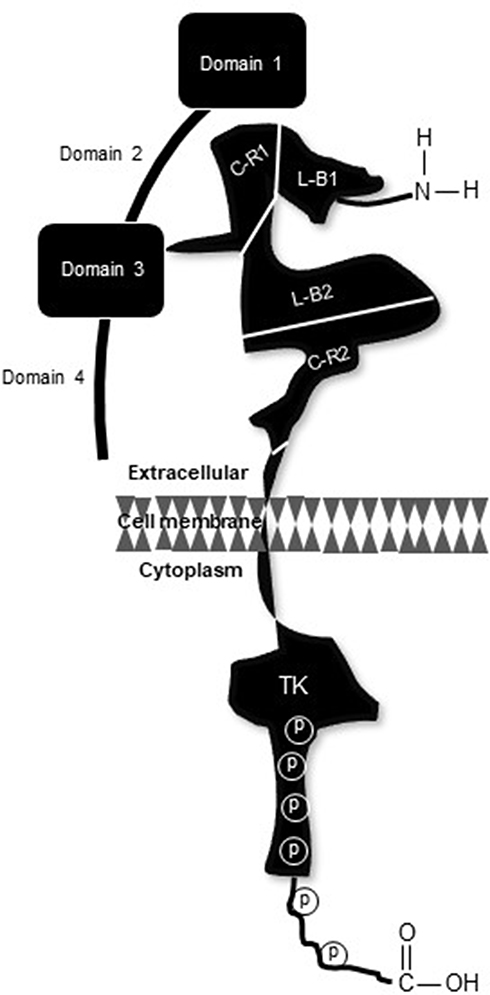 |
Figure 1 Pictorial rendition of p185HER2. Distal to the amino terminus, the extracellular region consists of four distinct sectors; two ligand-binding domains (L-B1 and L-B2) and two flanking cysteine-rich domains (C-R1 and C-R2), join to a short membrane-spanning region which connects with the intracellular catalytic kinase domain (TK). Sites of receptor phosphorylation are embedded in the TK region and along the carboxy terminus (denoted by P). |
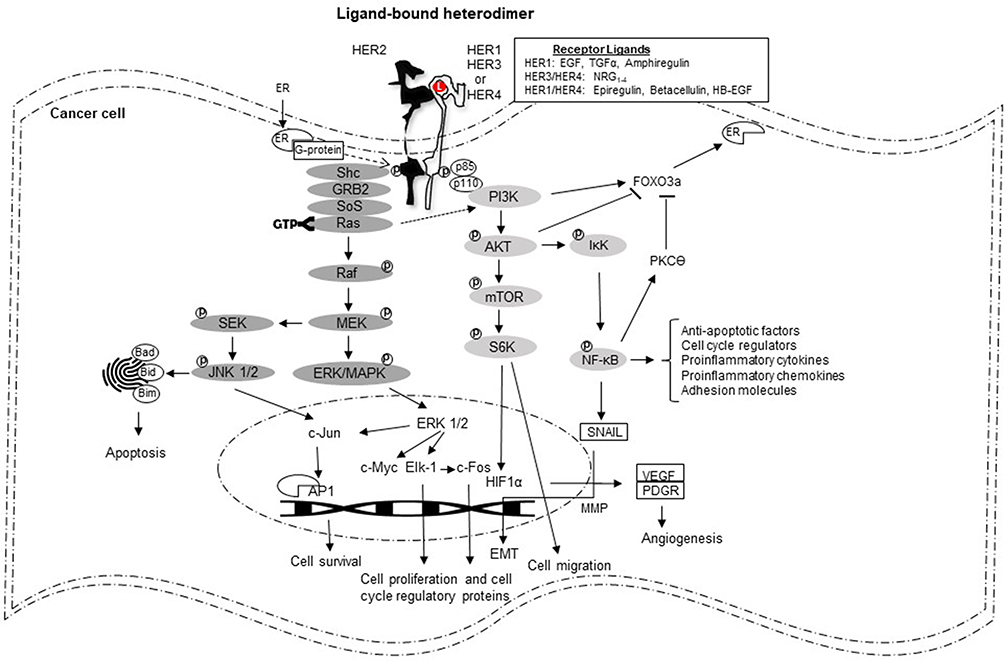
Though not absolute, cell surface receptors are biological versions of yin and yang. Anatomically, these structural components have key roles in transducing external stimuli into variable intramural messages (Figure 2). Deemed pivotal for normal physiological function, the binate character of the receptor could also be pernicious. For example, signaling initiated through normally expressed HER2 nurtures growth and development of mammary epithelium9 contrasts the malignant edict of overexpressed HER2 in breast cancer. Importantly, rather than the presence of the receptor alone, which does not distinguish this tumor subtype, HER2 overexpression (immunohistochemistry score of 3+) or/and gene amplification (detectable by fluorescence in-situ hybridization) is/are the standard diagnostic criterion/criteria.10 Furthermore, the relevance associated with receptor overexpression has been validated in three important ways: first, predictive and prognostic utility of this unique molecular feature;11,12 second, biological and clinical identity as a major tumor driver;13 and third, near absolute inevitability of resistance developing in metastatic disease. The last consideration highlights the obligation of scientists to develop additional pharmacologic choices, especially since HER2 remains a clinically operational target.14

Therapies
Neu-Interventions
Nearly a quarter century ago, trastuzumab was initially approved for the treatment of HER2+ mBC. Since this milestone achievement, the continued search for new products is related primarily to intrinsic, or subsequent development of, resistance as well as tumor progression in both advanced and early disease settings. Embedded in an expanded discussion of all the approved agents are the landmark clinical trials that influenced current treatment guidelines.
Antibodies
Not only did early laboratory studies indicate that HER2 could recast normal cells into cells exhibiting a malignant phenotype but also high-level expression of the receptor on approximately 20% of breast cancers culminated in the development and approval of trastuzumab (Table 1). The humanized antibody was configured with a complementarity determining region (CDR) with site-specific avidity in C-R2 (juxta-membranous domain IV) of the extracellular portion of the receptor (Figure 1). That no endogenous ligand manifests an affinity for the receptor may be partially attributable to HER2 alone. Using a crystalline construct to analyze the receptor revealed a couple of important features of the protein. First, impediment of ligand binding appeared to be associated with the misaligned “closed” arrangement of HER2, an atypical posturing that contrasts the symmetrically encrypted “open” configuration of the other three members of the family;15 and second, constitutional flaws in the CDR seemed to be related to remnants of the native amino acid sequence.16 Because of these findings, the target epitope for trastuzumab is likely different from the original ligand-binding locus.
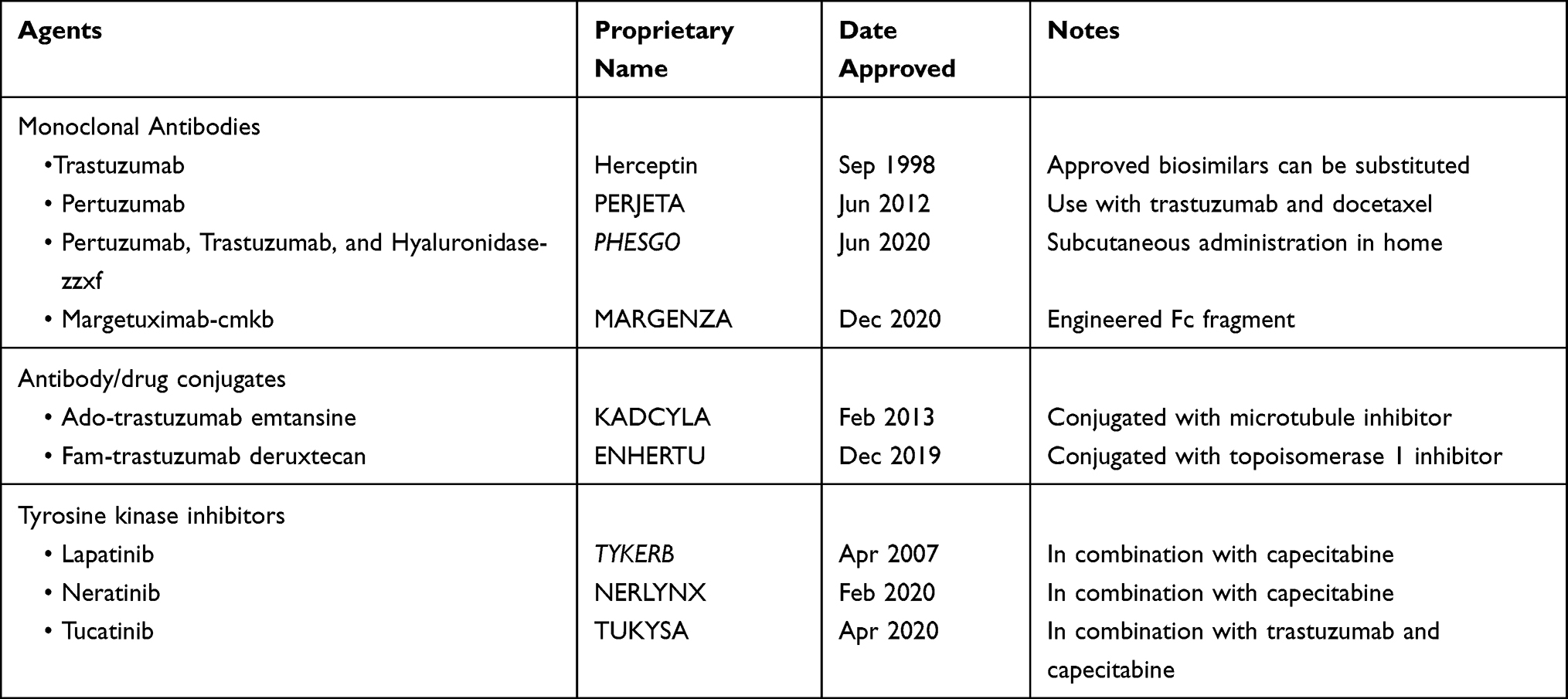 |
Table 1 FDA-Approved HER2-Directed Therapies |
Arguably the most important clinical trial to assess the efficacy and safety of trastuzumab involved women with previously untreated advanced HER2-positive breast cancer.13 Four hundred and sixty-nine patients were randomized (1:1) to receive standard chemotherapy with (n=235) or without (n=234) the antibody. Standard chemotherapy consisted of a combination of an anthracycline plus cyclophosphamide or paclitaxel monotherapy depending on prior adjuvant anthracycline treatment. Primary end point was time to disease progression (TTP); secondary endpoints were overall response rate (ORR), duration of response (DOR), and overall survival (OS). Analyses of the data indicated all the endpoints favored the trastuzumab-containing arm including a longer median TTP (7.4 vs 4.6 months; p<0.001), higher ORR (50% vs 32%, p<0.001), and longer median DOR (9.1 vs 6.1 months; p<0.001) and median OS (25.1 vs 20.3 months; p=0.046).
Significant also was the unexpectedly high rate of adverse cardiac events observed in this study that, in retrospect, was conspicuously absent in earlier phase clinical trials.17,18 Even though the precise biological mechanism(s) which mediate this adverse effect is not completely understood, selected data are available that provide some insight into the toxic reaction. Notably, the presence of HER2 is not restricted to cardiac tissue in adults, but the initial surfacing of the receptor coincides with cardiac morphogenesis in the fetus.19 Differences in the location of receptor expression in adult and fetal hearts provide a plausible explanation for the recognized myocardial damage associated with trastuzumab therapy. The appearance of HER2 in transverse (t)-tubules of mature ventricles secretes micro-passages that act as a conduit to store calcium ions in the sarcoplasmic reticulum.20 This functionally ideal communication enables integration of calcium flux with electrical coupling of excitation and contraction.21 Not only do these findings infer that HER2 signaling has a kinetic influence on the metronomic mechanics of the heart but also renders a physiological explanation for the reported cardiac injury.
Humanized like trastuzumab but unlike its predecessor, pertuzumab recognizes an epitope located on C-R1 (domain II) of HER2 (Figure 1). This discovery reemphasizes the issue related to the receptor’s ligand-binding configuration. Construct analyses of HER2 disclosed that contiguity of L-B1 and L-B2 (ie, the antibody’s prime tethering site) “extends” the receptor’s conformation, which limited access, and possibly contributed, to hindering identification of the receptor’s native ligand(s).15 Furthermore, this unique structural alignment may also be the premise upon which HER2 appears to be the “favored” heterodimeric partner, particularly of HER3.22 Interestingly, pertuzumab’s distinct binding site appears to affect HER2 in a manner divergent from trastuzumab.23 Attachment to domain II modifies protein symmetry causing steric hindrance, which obstructs dimerization with other receptors. This effect may be therapeutically significant because the ligand-bound dimeric partner transactivates HER2; and the aggregate inhibitory upshot may exceed that of trastuzumab.24
Results of a randomized Phase 3 clinical trial involving patients with HER2+ mBC led to the drug’s approval by the FDA (Table 1).25 Of the total 808 subjects enrolled, docetaxel and trastuzumab were combined with placebo (control) or pertuzumab in a 1:1 randomization. Independent committee assessment of progression-free survival (PFS) was the primary study endpoint; secondary outcomes of interest included investigator-appraised PFS, OS, ORR, and safety profile. With a follow-up of approximately 19 months, median PFS were 12.4 months (placebo group) versus 18.5 months (pertuzumab group) (HR, 0.62; 95% CI, 0.51 to 0.75; p<0.001). Early analysis of the data also indicated 23.6% (96) of the subjects receiving placebo had died compared to 17.2% (69) of patients receiving pertuzumab (HR, 0.64; 95% CI, 0.47 to 0.88). At the time of this assessment, the 165 deaths, which accounted for less than half of the prespecified total number of events for the final analysis, precluded any conclusive statement. However, analysis of more mature data with a median follow-up of approximately 8.25 years in both groups showed OS was 40.8 months compared to 57.1 months, in the control and pertuzumab arms, respectively (HR, 0.69%; 95% CI, 0.58 to 0.82; p<0.001); and with accumulated 235 events (58% of the total number), the 8-year landmark OS rates were 23% (95% CI 19 to 28) in the placebo group and 37% (95% CI 31 to 42) in the pertuzumab group.26 Notably, the side effect profiles did differ in certain aspects. Dermatologic effects such as dry skin and rash, and gastrointestinal events including diarrhea and mucositis occurred more frequently with the two antibodies. A near two-fold increase in febrile neutropenia was also observed in the pertuzumab group. Although uncommon, cardiac dysfunction (any grade toxicity) was reported nearly twice as frequently among those receiving placebo compared to pertuzumab. The final analysis of this milestone trial demonstrated that the addition of pertuzumab resulted in an unprecedented improvement in median OS that approached 5 years.
Novel in an esoteric way, margetuximab is a chimeric monoclonal antibody, which features an engineered fragment in the Fc region of the immunoglobulin. The rationale for this structural modification was based on the premise that a major part of the antibody’s anti-tumor effect results from Fc-indirect cellular cytotoxicity or ADCC.27 Although antibody-dependent cell-mediated cytotoxicity is contingent upon interactions between site-specific Fc regions with Fcγ receptors (FcγR), the process is complicated by the presence of activating and inhibitory receptors expressed on innate immune effector cells. Sophisticated laboratory studies indicated that glycoengineering changes of several amino acids in the constant region could increase the antibody’s affinity to activating receptor CD16A (FcγR) and decrease its affinity to inhibitory receptor CD32B.28 Of the two Fc mutants generated with the most pronounced activity, one containing five amino acid changes was able to achieve a higher level of ADCC and tumor lysis at a lower drug concentration.
Margetuximab’s discreet Fc motif is based on the findings noted above. The modified immunoglobulin is similar to trastuzumab in terms of isotype and epitope specificity. And like trastuzumab tumor cell killing is accomplished in an Fc-independent manner. The focal difference between the two antibodies regards margetuximab’s engineered affinity to the activating (rather than inhibitory) receptor expressed on components of the innate and adaptive immune response targeting HER2. Proof of biological principle based on the hypothesis that preferred interlinkage between Fc and FcγR would be clinically beneficial was assessed in a phase 3 clinical trial.29 Eligibility was restricted to subjects with progressive disease after >2 HER2-directed therapies. Enrolled patients were randomized to margetuximab or trastuzumab plus single-agent chemotherapy of investigator's choice. Primary study endpoints were PFS and OS. Of the 536 evaluable patients, 270 and 266 received margetuximab and trastuzumab, respectively. The first of two pre-planned blinded analyses of the data showed a modest comparative PFS benefit favoring margetuximab (5.8 months vs 4.9 months; HR, 0.76; 95% CI, 0.59–0.98;p=0.03). At the second interim analysis, an improved ORR was observed with margetuximab (25% vs 14%) (p<0.001). Safety profiles were similar except for an increased incidence of infusion-related reactions with margetuximab. This distinctive new agent was approved in December 2020 for advanced HER2-positive breast cancer in patients who have received at least one trastuzumab-containing regimen.
Despite the positive results, some may argue that the enthusiasm of this engineering feat could be tempered by the study's detail that all evaluable subjects had disease progressing on trastuzumab and, nearly all, ado-trastuzumab emtansine. Hence, it is possible that the primary study endpoints were prematurely skewed against those randomized to the trastuzumab group. Countering this argument are published data demonstrating that disease progression may not be due solely to loss of antibody activity but rather to the chemotherapeutic agent(s) used in combination.14 Nonetheless, this issue can be resolved with a clinical trial comparing the two antibodies in treatment-naïve subjects with HER2+ mBC.
PHESGO, the proprietary name for a fixed-dose combination of pertuzumab and trastuzumab with hyaluronidase-zzxf, incorporated in the formula to facilitate absorption following subcutaneous administration. Approvals of this product were essentially non-inferior profiles, which hinged on efficacy and safety outcomes, with the exception of higher administration-related reactions with PHESGO, observed in patients with non-metastatic, HER2-positive breast cancer (Table 1).30
Antibody-Drug Conjugates (ADCs)
One of the most important concepts related to the development of ADCs was the lack of tumor selectivity associated with chemotherapy. Coupling target specificity with the observation that trastuzumab’s modest anti-tumor effect could be substantially improved by the addition of a cytotoxic agent led to the idea that antibodies could be used to deliver chemotherapy rather selectively to tumor cells. The development of a novel therapeutic compound known as T-DM1 or trastuzumab-emtansine appeared to overcome certain limitations associated with targeted-antigen expression, tumor cell uptake mechanisms, and cumbersome linkers used in the conjugation process.
Perhaps, the most definitive results emerged from the landmark phase 3 clinical trial involving patients with disease progressing on front-line trastuzumab-based therapy (Table 1).31 Randomized coequally to T-DM1 or the current second-line combination of lapatinib and capecitabine, patients in each group were treated until disease progression or intractable toxicity. Independently assessed PFS, OS, and safety were the principal endpoints. A total of 978 patients of the 991 enrolled, with a median duration of follow-up of approximately 12 months, were subject to data analyses. Among those receiving T-DM1, the median PFS was 33% longer than the comparative group, 9.6 months vs 6.4 months; HR, 0.65 (95% CI, 0.55 to 0.77; p<0.0001). Although not evaluable initially, at the second interim analysis median OS crossed the pre-specified efficacy-terminating border supporting the ADC, 30.9 mo vs 25.1 mo; HR, 0.68 (95% CI, 0.55 to 0.85; p<0.001); ORR also favored T-DM1 (43.6%, vs 30.8%; p<0.001). Grade ≥ 3 AEs observed more frequently with T-DM1 included thrombocytopenia and increased liver function tests. Not unexpectedly, the incidence of diarrhea and hand-foot syndrome was higher in those treated with the capecitabine-containing combination.
[Fam-] trastuzumab deruxtecan is the second HER2-targeted ADC. Coupling the signet HER2 antibody with a topoisomerase I inhibitor, the newer ADC was granted accelerated FDA approval in December 2019 based on the results of the Phase 2 DESTINY-Breast01 trial.32 In this multicenter, single-arm trial, 184 patients with previously treated HER2-positive metastatic breast cancer received the studied-compound every 3 weeks until disease progress or unacceptable toxicity. The primary efficacy outcome was confirmed ORR and response duration assessed by independent central review. At the time of publication, 60.3% (95% CI: 52.9, 67.4) of the participants had an objective response to treatment. Though most responses were partial (56%), a few (4.3%) were complete; median duration of response approached 15 months (95% CI: 13.8, 16.9). Of interest, objective responses were achieved in 36 of 56 subjects previously treated with T-DM1. While seemingly modest, what made these outcomes particularly noteworthy was the median number of therapies (ie, 6) the subjects received prior to study enrollment. Nonetheless, clinical benefits did come with a moderate degree of toxicity, including bone marrow suppression affecting all cell lineages, gastrointestinal side effects, such as nausea, vomiting, and diarrhea, and constitutional symptoms, such as fatigue, all of which had reported frequencies of at least 20%.
Because of fatal outcomes due to interstitial lung disease (ILD), the approval of this compound came with a note of caution. While this toxic event poses an important risk to patients, the boxed warning came prior to a formal assessment of this adverse effect. To better characterize the reactive ILD, a refereed audit of patients who received the ADC was performed.33 Of the total population, any-grade ILD was observed in approximately 16% of the 879 subjects included in the pooled analysis. The findings of an independent adjudication committee indicated that 78% of all cases were grade 1 or 2 with a median time to onset of 5.5 months. With 28% of patients remaining on treatment for more than 12 months, the committee also observed that the risk of developing ILD actually decreased over time. The committee also identified several risk factors that have not yet been validated. Even so, the list includes ethnic Japanese, dose >7.4 mg/kg, baseline oxygen saturation <95%, presence of moderate/severe kidney dysfunction or lung comorbidities, and time from initial cancer diagnosis >3.9 years. While these findings do not marginalize the toxicity, the results provide further clarity on treatment-associated ILD in addition to management guidelines that have been implemented.
Small Molecule TKIs
Lapatinib is a reversible, bi-specific inhibitor of EGFR and HER2. Results of timed crystalline scanning of both lapatinib/EGFR and erlotinib/EGFR disclosed dissimilar dissociation constants for each inhibitor; separation occurred slower with lapatinib translated into longer inhibition of receptor phosphorylation in tumor cells.34 Even though outcomes of similar quantitative research involving the lapatinib/HER2 complex are not available, some evidence suggests that catalytic activation of the receptor is comparably suppressed.35
Lapatinib is the second approved agent to demonstrate significant clinical benefits in patients with HER2-positive, advanced breast cancer progressing on trastuzumab (Table 1).36 Several salient details of this pivotal phase 3 trial should be highlighted. First, the results of a predetermined interval analysis of TTP and PFS data indicate a statistically meaningful difference among patients who received the lapatinib-containing regimen compared to capecitabine alone led to early termination of the trial. Second, the lower incidence of brain metastasis suggests CNS penetration and antitumor activity of the TKI. Third, tumor expression of EGFR did not appear to influence the drug’s clinical efficacy since subject eligibility was not dependent on tumor cell expression of the receptor. Fourth, debunking of the belief that significant benefits in the lapatinib arm were an artifact due to trastuzumab given in the previous eight weeks.37
Neratinib was initially approved for extended adjuvant treatment following adjuvant trastuzumab-based therapy.38 Subsequently, the agent received an FDA endorsement for use as a 3rd line anti-HER2 therapy following the release of data from the NALA trial (Table 1).39 Treatment-related eligibility for this randomized phase 3 clinical trial was restricted to patients who had received at least 2 prior anti-HER2-directed therapies. Co-primary endpoints were PFS and OS. Six hundred and twenty-one subjects were randomized to receive capecitabine with either neratinib or lapatinib. An improvement in PFS was observed among subjects treated with neratinib – compared to lapatinib-containing arms (HR, 0.76; 95% CI, 0.63–0.93; p=0.0059). At 12 months, PFS was found to be 29% versus 15% in the neratinib and lapatinib arms, respectively. Median OS of 21 months versus 18.7 months in the respective arms was not statistically different. Overall response rates and duration of responses were modestly improved with neratinib. The most significant treatment-emergent adverse effect was diarrhea in both arms, although grade ¾ toxicity was two-fold higher with neratinib.
The improved clinical outcomes observed in this study suggest that neratinib is superior to lapatinib. If so, it would be appropriate to examine what may be the underlying bases that distinguish the two TKIs. Studies using breast cancer cell lines demonstrated that neratinib inhibited phosphorylation of not only HER1 and HER2 but also HER4.40 When tested, neratinib was also able to suppress the proto-oncogene cellular (c)-Src though to a lesser degree. Nonetheless, the potential ramifications against tumor cells are numerous as the c-Src kinase is downstream of, and activated by, EGFR.41,42 In addition, c-Src signaling contributes to angiogenesis as well as tumor cell proliferation and survival.43–45 Another modification, incorporation of a chemical moiety known as Michael Addition, enables neratinib to bind irreversibly and promotes lasting receptor inhibition, which may be responsible for the greater potency of neratinib.46 In addition, compared to the steadfast bond resulting in irreversible inhibition of neratinib’s targeted proteins, laboratory studies showing the slow recovery of enzyme activity suggest that lapatinib does not form stable covalent bonds.34
Although both neratinib and lapatinib have been shown to reverse multi-drug tumor resistance (MDR) attributable to overexpression of ATP-binding cassette (ABC) elements, the MDR reversal effect of neratinib is quite unique. Instead of decreasing the expression of membrane-bound ATP transporters, blocking downstream phosphorylation of AKT, or facilitating intracellular translocation of the ABC protein, neratinib effects MDR reversal by directly inhibiting the efflux function of ABCB1.47,48
Two months after neratinib became available for clinical use, a third small-molecule inhibitor was approved as a second- or third-line treatment for advanced HER2-positive breast cancer (Table 1). The application of the newest agent, tucatinib, in conjunction with capecitabine and trastuzumab even included patients with brain metastasis. In contrast to lapatinib and neratinib, tucatinib is HER2-selective. Its repressive effect also translates to downstream inhibition of AKT and ERK phosphorylation.49 Basis for approval were results emanating from the HER2CLIMB trial.50 All the subjects enrolled, including those with metastasis to the brain, had received two of the current frontline HER2-directed therapies. Randomized patients were treated with trastuzumab and capecitabine with or without tucatinib. A key study endpoint was PFS; outcomes concerning PFS among those with CNS disease, OS and safety were also assessed. Analysis of data at one-year follow-up indicated a difference in PFS favoring the tucatinib group, 33.1% vs 12.3% (HR, 0.54; 95% CI, 0.42 to 0.71; p<0.001). At this same time point, PFS in subjects with brain metastasis was 24.9% compared to 0%, again favoring the tucatinib arm (HR, 0.48; 95% CI, 0.34 to 0.69; p<0.001). At two years, the OS rate was 44.9% and 26.6% in the tucatinib and placebo groups, respectively. The most common adverse events observed among those receiving tucatinib were relatively severe diarrhea and transaminitis (both grade ≥ 3), as well as hand-foot syndrome, nausea, vomiting and fatigue.
Although not directly comparable with other clinical trials, two aspects of HER2CLIMB present an opportunity for reasoned speculation regarding the three TKIs. First, that treatment-related AEs lead to discontinuation of therapy, lower in this study (~6%) compared to the NALA trial (16–18%), suggest that multi-kinase targeting TKIs carry extra risks with seemingly minimal additional therapeutic benefit. This consideration is further supported by the fact that the capecitabine dosing regimens were identical in both trials. Second, the longer median PFS with two HER2-directed agents (7.8 months) compared to monotherapy (5.6 months) infers that inhibiting the receptor’s extra- and intra-cellular domains may have an additive effect. This perspective is bolstered by the observation of PFS benefit occurring across the entire study population in the tucatinib arm.
Since trastuzumab’s approval 20 years ago as the first HER2-directed therapy, eight additional products (excluding biosimilars) are now available. And even after numerous clinical trials have been conducted, the original antibody continues to maintain its preeminent position in the management of metastatic HER2-positive breast cancer. Therefore, and not surprising, instead of a dramatic change in the treatment landscape, each approved product has garnered sufficient credence to establish a position, some flexible and likely temporary, in the hierarchy of the current treatment stratagem (Table 2).
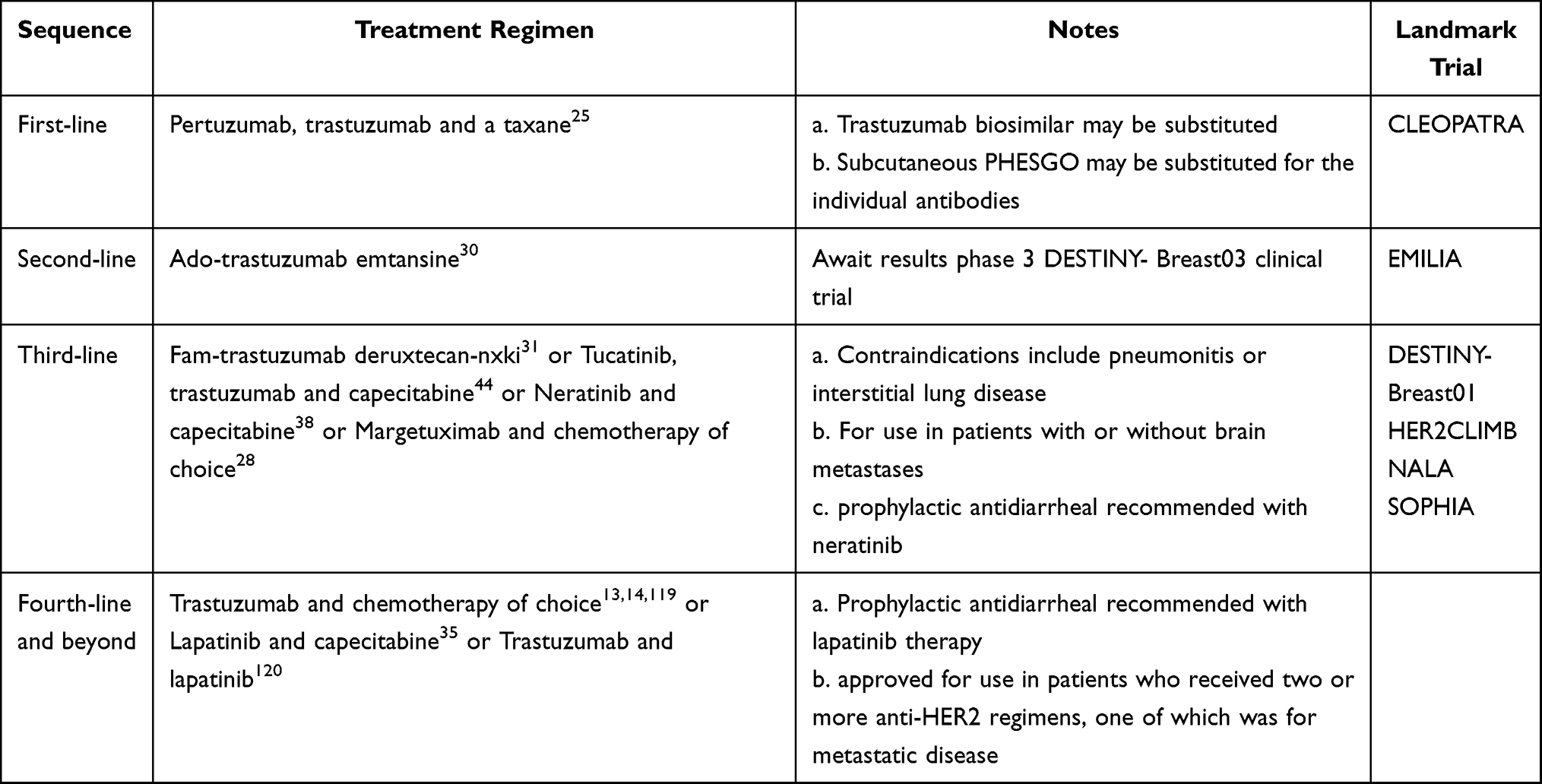 |
Table 2 Evidence-Based Treatment Recommendations for Metastatic HER2-Positive Breast Cancer |
Maneuvering Forward
Although the focus has been on the HER2-rich subtype, receptor overexpression also occurs in one of the luminal B subtypes. In ER+, HER2+ metastatic breast cancer, pertuzumab and trastuzumab plus an aromatase inhibitor may be a first-line option in subjects without significant visceral compromise.51 In patients who are candidates for cytotoxic chemotherapy, taxanes can be continued in responsive disease and in the absence of significant side effects. In the event of treatment-limiting toxicity, particularly neuropathy, combined HER2 blockade can be continued with the addition of appropriate endocrine therapy.
A number of ongoing phase 3 clinical trials were designed to further evaluate two approved ADCs. Destiny-Breast02 is a comparative trial of [Fam-] trastuzumab deruxtecan (T-DXd) and treatment of investigator’s choice for metastatic HER2-positive breast cancer in patients previously treated with ado-trastuzumab emtansine (T-DM1). However, the most anticipated trial appears to be Destiny-Breast03 which compares [Fam-] trastuzumab deruxtecan against ado-trastuzumab emtansine as second-line therapy in subjects who received prior therapy with trastuzumab and a taxane. Still, another, though unique, clinical trial (Destiny-Breast04) compares T-DXd and physician’s choice of treatment in patients with metastatic, HER2-low expressing breast cancer who were previously treated with chemotherapy. This study attempts to define a new HER2 population who may benefit from the targeted ADC by including tumors with IHC scores of 1+/2+ or FISH-negative HER2 expression. Another notable phase 3 clinical trial (HER2CLIMB-02) is a placebo-controlled study of tucatinib in combination with TDM1 for patients with unresectable locally advanced or metastatic HER2-positive breast cancer.
Currently in progress also are several early phase trials in subjects with advanced HER2-positive breast cancer involving the brain. Some of the more engaging studies include a genomically guided trial targeting actionable alterations in neurotrophic tyrosine receptor kinase, ros oncogene 1, and cyclin-dependent kinase or PI3K/Akt; intraventricular infusion of autologous chimeric antigen receptor T cells targeting HER2; atezolizumab in combination with pertuzumab and high-dose trastuzumab; and a comparison of T-DM1 alone versus T-DM1 plus metronomic temozolomide in secondary prevention of brain metastases following stereotactic radiosurgery. Two other therapeutic agents in initial clinical trials are bi-specific antibodies, which target HER2 and CD3 or HER3 and neuregulin 1 (NRG1), one of the HER3 binding ligands.
While some of these ongoing clinical trials will have a significant impact on disease management, recently accumulated evidence demonstrates only modest therapeutic benefit of immune checkpoint inhibitors in PD-L1-positive TNBC.52,53 Part of the explanation for the lower priority of conducting clinical trials of checkpoint inhibitors in breast cancer may relate to the disease being largely hormone receptor-positive. Luminal breast cancers also demonstrate lower tumor PD-L1 expression and mutational burden, manifestations of which suggest a lack of immunogenicity.54–56 Nevertheless, if these markers do correlate with response, benefit could still be gained by enriching the study population to include only subjects with tumors expressing these markers regardless of molecular cancer subtype. Very recent clinical trial data appear to add some credence to this argument.57 The phase 2 TAPUR study involved patients with previously treated metastatic breast cancer of any subtype manifesting high tumor mutational burden. All subjects were treated with monotherapy PD-1 inhibitor. Primary endpoint was disease control (DC) defined as objective response or stable disease of at least 16 weeks of duration. The study used Simon’s two-stage design with a maximum recruitment of 28 evaluable subjects. After fulfilling the requirement of two or more of the first 10 patients in the first stage achieving DC, 18 additional subjects were enrolled. Disease control was achieved in 37% (95% CI, 21 to 50) of the patients. Median PFS was 10.6 weeks (95% CI, 7.7 to 21.1); median OS was 30.6 weeks (95% CI, 18.3 to 103.3).
Additional evidence supporting the role of checkpoint inhibitors includes an early phase clinical trial of combined PD-1 inhibitor and trastuzumab in patients with trastuzumab-resistant advanced HER2-positive breast cancer. Results of the PANACEA trial indicated that in the subgroup of patients with PD-L1-positive disease and >5% TILs in the metastatic lesion, the ORR was 15%; the disease control rate was 23% with a median duration of 11.1 months. Of interest also, no responses were observed in the absence of PD-L1 expression. The importance of PD-L1 appeared to correlate with PFS, estimated 13% at 12 months in PD-L1-positive and 0% in PD-L1-negative patients; estimates of one-year survival were 65% and 12%, respectively.58
Challenges
Neu-Resistance
Although extensive research has revealed not only the presence but also the influence of HER2 in breast cancer, it is still possible that only the façade, and some dynamics, of the receptor has been exposed. Conversely, the evolution to, and development of, resistance is nearly absolute.59 Whether one accepts the theory or not, tumor resistance has an aura of Darwinism; and like cancer, resistance appears to be an evolutionary process, wherein genomic instability sows the seeds of natural selection and formative retention of traits that favor tumor cell survival and growth.
Though speculative, a pragmatic attempt is made at deconstructing some of the mechanistic or molecular bases of HER2-resistance. Perhaps the most-timely, and still puzzling, consideration relates to the immune checkpoints among which programmed cell death protein-1 (PD-1) and its ligand PD-L1 have been most prominent. Discovered within the past two decades,60,61 the mutual affinity between receptor and ligand is believed to be a major pathway of tumor cells that evade immune destruction; and hence, it could also have a considerable role in tumor resistance. Indeed, PD-L1 expression has already been correlated with poor clinical outcome in lung, kidney, and colorectal cancers.62,63 However, this association has not been consistently observed. While heterogeneously expressed among breast cancer subtypes, PD-L1 is generally correlated with intra-tumoral lymphocyte infiltration and poor-prognostic disease features including the HER2-enriched molecular subtype.64 Surprisingly, and somewhat counterintuitively, are published reports of PD-L1 expression being associated with a favorable prognosis for breast cancer.65–67 Still, this finding is not as discordant as it appears and may, in part, be related to the source of ligand expression. Embedded in formalin-fixed tumor samples are a number of immune cellular components, such as a bevy of tumor-infiltrating lymphocytes (TILs), macrophages, and antigen-presenting cells, which also express PD-L1.68,69 While the use of paraffin-embedded tissue is not novel, careful discrimination between cancer and immune cell expression of checkpoint proteins is. Because ligand expression is not restricted to tumor cells, “confirmation” of PD-L1-positivity could obscure the anti-tumor effects of certain TIL subsets such as those positive for lymphocyte-activating gene 3 and CD4+ regulatory cells (Tregs) which could partially explain the favorable breast cancer outcomes.70 Furthermore, effector TILs also express high levels of several proinflammatory cytokines as well as the serine protease granzyme B. Among these, interferon-gamma has been shown to be a potent inducer of PD-L1 expression.71 If the aforementioned concepts are valid, PD-L1 expression on immune molecules may also be an indicator of a robust immunologic response.
Although HER2 may not be the most dominant family member, it appears to be the most perplexing. One consequence of receptor overexpression is the emergence of homodimers.15 While this dimeric couple possesses constitutive tumor-promoting activity, the development of tumor resistance may not be due solely to pairing of the receptor but to the formation of an unmatched HER2 dimer (Figure 3).72 For instance, laboratory studies demonstrate that ligand-activated HER3 and HER1 are key components, which influence receptor phosphorylation as well as recruitment of proximal proteins.73 These data may demystify the finding that neither HER2 nor HER3 monomers are able to activate downstream kinases alone, yet melding of the two receptors endows the heterodimer with potent tumorigenic capability.74 Likewise, mitogenic signals originating from dimerized EGFR and HER2 have also been reported to be more robust than either homodimer.75 The logic behind the development of agents that inhibit the activation of EGFR and HER3 was based on these findings; uncertainty still exists regarding the role of HER4 in the tumorigenic process.
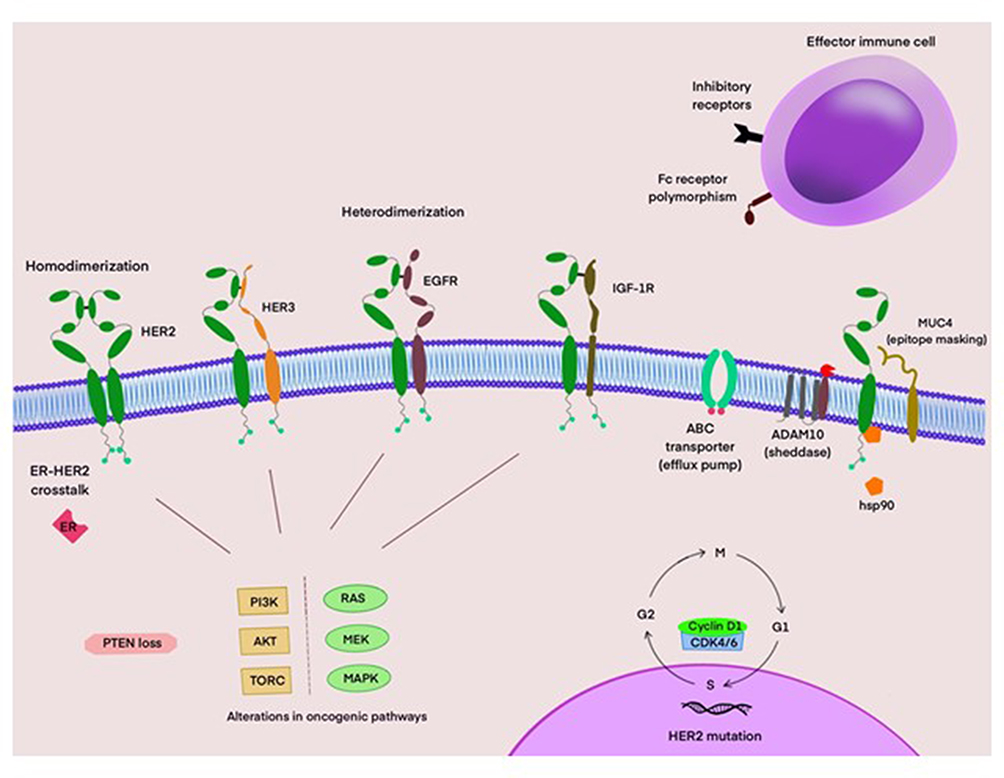 |
Figure 3 Putative mechanisms of resistance to HER2-directed therapies. HER2 homo- and hetero-dimers activate key signaling pathways. Mutations involving the PI3K/AKT/TORC and RAS/MEK/MAPK pathways, as well as loss of PTEN promote tumor cell growth and survival. Certain Fc receptor subtypes, such as FcγRIIB, or polymorphisms lower antibody binding affinity. HSP90 impairs endocytosis of the trastuzumab-HER2 complex. Upregulation of inhibitory receptors in the tumor microenvironment impair anti-tumor immune responses. Cyclin D1-overexpressing tumors demonstrate reduced responsiveness to trastuzumab-based therapy. Overexpression of cyclin D1 reduces responsiveness to trastuzumab-based therapy. ADAM10 cleaves the extracellular domain of HER2 resulting in a constitutively active truncated receptor. The MUC4 protein interferes with trastuzumab binding through epitope masking. Drug efflux pumps like ABC transporters can also reduce efficacy to HER2-targeted therapies. |
HER2 resistance may be intrinsic to the receptor itself as a number of mutations have been sequenced. Mutations in the most-studied kinase domain appear to be localized to the ATP-binding site, amino (N) terminal region, and carbohydrate (C) terminal lobe and hence, may alter receptor activity by different mechanisms. Nonetheless, those found to be activating mutations conferred not only more aggressive tumor features but also resistance.76 Interestingly, cells resistant to lapatinib were sensitive to the inhibitory effects of neratinib.77 While the characterization of gene mutations is not necessarily novel, what is notable about HER2 is the detection of somatic mutations in both gene-amplified and non-amplified breast cancers.78 If the activating mutations are consensus tumor drivers, the clinical significance of the previous finding may enhance drug selection in HER2-amplified tumors as well as provide more effective treatment options for HER2-“negative” breast cancers.
Resistance to the receptor may also be structurally related as intact HER2 (p185ErbB2) can be truncated to a shorter but molecularly detectable protein (p95ErbB2) with constitutive kinase activity.79 Subsequent studies identified a disintegrin and metalloprotease (ADAM) 10, an EGFR ligand sheddase, as the enzyme that severs the ectodomain of HER2.80 The cleaved receptor is remarkable for two additional reasons. One, p95ErbB2 is the preferred dimerization partner of HER3; and two, due to loss of the binding region, the truncated receptor is not susceptible to trastuzumab-induced inhibition of its catalytic activity.34 While increased serum concentrations of the cleaved external domain (CED) have been linked to poorer therapeutic outcomes as well as lymph node involvement,81 this finding may also have predictive value for the use of small-molecule TKIs. The latter idea also supplies a reasonable, though incomplete, explanation for the effectiveness of second-line lapatinib in patients with EGFR-expressing tumors and high serum levels of the CED.
Insulin-like growth factor-1 receptor (IGF-1R) may be another mechanism of HER2 resistance and is supported by laboratory studies and clinical outcomes data. Proof of the former is supported by in-vitro experiments with several human breast cancer cell lines that overexpress both receptors. While trastuzumab alone had only a modest effect on cell proliferation, dual blockade of HER2 and IGF-1R resulted in a three-fold greater reduction in tumor growth.82 Clinical evidence gleaned from locally advanced HER-positive breast cancer studies also shows an indirect correlation between positive IGF-1R expression in tumor cells and poorer response outcomes with neo-adjuvant chemotherapy plus trastuzumab.83 Contrary to the perception that dimer formation is restricted to HER-family members, these data suggest that other receptors also have the capacity to effect the activation status of HER2.84 In addition to receptor phosphorylation, HER2 resistance may stem from IGF-1R-activated kinase complexes, such as cyclin/cyclin-dependent kinase 2 in a manner different from HER2.85 These data infer, but do not assure, that inhibition of both receptors will ultimately translate into therapeutic benefit. This element of reservation is based on molecular and clinical characteristics of the respective genes. The authors’ inherent skepticism is supported by one pivotal advance: the utility of gene amplification. In particular, multiple copies of the HER2gene, which has been successfully used as a guide for HER2-directed therapy, are not matched by any companion feature involving IGF-1R.
Yet another binary arrangement of HER2 that could affect the development of resistance involves the estrogen receptor, specifically ERα. Though not a transmembrane receptor, the link between the two receptors may have “evolved”, not in an evolutionary manner, but rather in conjunction with genetic conservation of a liaison reliant on bidirectional flow of signals that can ultimately affect each other.86 For example, upregulation of the ligand heregulin has been shown to accompany hormone-depriving endocrine therapies.87 Conversely, compelling experimental data demonstrate how HER2 may be able to promote endocrine resistance in hormone-dependent breast cancer.88 Additional evidence supporting the validity of the last-mentioned finding is the observed hormone resistance in luminal B tumor cells that co-express both ER and HER2.89 That these findings have translational merit is the observation from two late phase clinical trials showing significantly improved PFS in subjects with luminal B (HER2-positive) in the group receiving therapies targeting estrogen synthesis and HER2 compared to estrogen deprivation therapy alone.90,91
However, the reciprocal also appears to be true. For example, resistance to anti-HER2 therapies may result from reactivation of the ER-signaling pathway. This has been demonstrated in animal models and human tumors.92 Even though an inverse relation between ER and HER2 expression in breast cancer has been observed in human tumors, effective blockade of the HER2 pathway has been reported to increase or restore ER levels in animal models. This development of HER2 resistance is related to restoration of the ER signaling pathway and is supported by the demonstration that ER inhibition leads to growth arrest and apoptosis in lapatinib- and lapatinib + trastuzumab-resistant cells.93
A number of cytoplasmic signaling complexes also appear to be associated with the deviant behavior of HER2-positive tumor cells. Constitutively activated kinases of the genomically altered phosphatidylinositol 3-kinase (PI3K) alliance may be another route to resistance. Even normal monitoring of cellular elements such as histone deacetylases can, enzymatically, effect a variety of cell outcomes including growth, differentiation, quiescence, and apoptosis.94 Tumor cells can also produce proteins such as those of the heat shock family in response to stress. Although basal levels of these proteins do exist, their latent roles become evident when overexpressed by stressful conditions. One demonstrable manifestation of the heat shock proteins that appears to coincide with HER2 resistance is by reversing receptor sensitivity to catabolism.95 However, what may be most enlightening about some of these cellular components is the unanticipated finding that they seem to dispute the belief that the tumorigenic process is directed exclusively by rogue proteins. In retrospect, focusing therapy only on aberrant targets has been a cleverly disguised escape for tumor cells.
Neu-Brain
Even though brain metastasis has been observed with all subtypes of breast cancer, encroachment of the central nervous system (CNS) is higher among subjects with HER2-positive compared to those with primarily hormone-sensitive disease.96,97 What makes the proclivity of brain metastasis intriguing relates not only to the receptor itself but also to the selective barrier tumor cells must infiltrate in order to gain access to the CNS. The relevance of brain metastases is further accentuated because of the impact of the disease on both quality and quantity of life.
There is biological evidence for HER2’s presumptive role in amplifying the metastatic potential of breast cancer cells. For example, overexpressed HER2 has been shown to promote TGFβ/SMAD-mediated activation of SNAIL, a family of zinc-finger transcription factors, including SNAIL, SLUG and SMUC, with repressive effects on E-cadherin and cytokeratin-18.98,99 The importance attached to decreased expression of these cell-adhesion molecules relates to their role in epithelial-to-mesenchymal transition or EMT, a process that confers tumor cells with enhanced migratory and invasive characteristics. Even so, it is clear that the HER2 phenotype alone is not the sole explanation for the CNS-migratory effects. This notion is strengthened by the observation that the incidence of brain metastases is also relatively common in TNBC.96 Regardless, this apparent disparity can be partially reconciled with the finding that breast cancers originally negative for HER overexpression were subsequently found to be HER2-positive when brain metastases were analyzed.100
An additional consideration relates to other HER family members. A report of various types of tumors, including breast cancer that metastasized to the brain, confirmed the presence of all HER receptors with overexpression of HER3 occurring most often.101 Furthermore, phosphorylated though catalytically impaired HER3, was observed more frequently in brain metastases compared to the respective primary tumors. This finding is perhaps less confounding because of two factors. First, a characteristic feature of the HER family is the substantial degree of heteromeric interactions;102 and second, central neuronal and glial cell expression of neuregulins, the cognate ligands for HER3.103 What is noteworthy is that overexpressed and activated HER3 was found even in non-HER2 gene-amplified TNBCs.
Another HER family member that appears to be important is EGFR (HER1). This supposition is supported by correlative clinical evidence showing significant differences in EGFR expression between brain metastases and primary tumors.104,105 However, more important than its mere presence are the machinations involving EGFR. Two of the growth factor receptor’s most relevant associations are heparanase (HPSE) and cyclooxygenase-2 (COX2). First, not only does HPSE activity correlate with migratory and invasive characteristics of tumor cells but also being adept at breaching the blood-brain and blood-tumor barriers. Indeed, a striking example of this attribute was the demonstrated proficiency in which transfected human breast-derived circulating tumor cells generated brain metastases in a murine model.106 Even though HPSE has been shown to increase EGFR phosphorylation, there is the intriguing possibility that enhanced HPSE-mediated colonization of brain tissue may occur independent of HER-signaling pathways by dynamically altering cell-adhesion molecules and critical cytoskeletal features.107,108 Second, the differential ability to colonize the brain relative to the lung may be due to a cluster of molecular abnormalities of which EGFR is one component. For example, overexpression of EGFR, HER2, Notch1 and HPSE as well as downregulation of epithelial barrier-protective proteins, such as periplakin or mitogen-activated protein kinase 13 have been observed in isolated circulating tumor cells from subjects with brain metastases.109 Molecular differences between primary breast tumors and invasive brain metastases suggest that the migratory, brain-selective cells are not merely replicative progenies of parental cells but rather unique genomic variants.
Although the focus on HER1 and HER3 does not in any way infer the exclusivity of these two receptors and their alleged pathological role in brain metastases, subversive crosstalk between HER1 and HER3 appears to initiate metastasis-promoting signaling in melanoma, another tumor with a predilection for invading central neuronal tissue.110 And despite their accepted importance manifested through dimeric coupling with ligand-less HER2, several ligand-dependent nuances are unique to the heteromeric crosstalk between EGFR and HER3. Compared to trans-activation of HER2 and HER3, which is dependent on the interactions emanating from the NRG-directed HER3/HER2 kinase dimer, the absence of NRG-induced clustering of HER3/EGFR oligomers results in phosphorylation of HER3 only.8 Juxtaposed to this finding is the virtual absence of EGFR phosphorylation in response to NRG binding to HER3. Indeed, the extent of dimerization appeared to mirror that of HER3 alone or small clusters of HER3/EGFR dimers. However, in the presence of EGF, higher order oligomeric complexes of the two receptors could account for the activation state of both receptors. This unexpected finding has two potentially profound implications. First, the contribution of each receptor may have the pathogenesis of brain metastases; and second, a mechanism through which tumor cells may escape the lethal effects of therapies targeting HER2.
Certainly not exhaustive, the preceding laboratory findings represent only a portion of what are still opaque mechanisms resulting in tumor cell egress into the CNS. Still, the plausibility of these processes is supported, in part, by clinical studies. For example, investigators of small phase 2 clinical trials with neratinib or lapatinib in combination with capecitabine reported objective responses ranged from 49% to 66% of the subjects with brain metastases.111,112 A couple of recently published late phase clinical trials add further support for small-molecule inhibitors in brain metastases. First, the NALA clinical trial, which compared neratinib or lapatinib, in combination with capecitabine, showed superior CNS outcomes with fewer interventions among subjects in the neratinib arm.39 Neratinib is distinguished from lapatinib in two ways. One, the former is an irreversible kinase inhibitor; and two, in addition to HER1 and HER2, neratinib also targets HER4. Whether these characteristics account for the differences in clinical CNS outcomes is uncertain. The second clinical trial involved an investigational, highly selective inhibitor of the HER2.50 The study randomized subjects to tucatinib or placebo, in combination with capecitabine and trastuzumab patients with metastatic breast cancer who previously received trastuzumab, pertuzumab, and trastuzumab-emtansine. Progression-free survival at 1 year among subjects with brain metastases was 24.9% and 0% among those in the tucatinib- and placebo-containing groups, respectively. While the CNS responses are inherently suggestive of efficacy, monotherapy studies with these TKIs showed much less activity against brain metastases. Furthermore, capecitabine has been reported to achieve significant though variable penetration into the brain and tumor cells.113 At the very least, these findings should moderate the certainty regarding the degree of central antitumor effects attributable exclusively to HER2-directed therapies.
Apart from its predictive and prognostic value, HER2 is less utilitarian as a biomarker of brain metastasis. The resulting dismal life expectancy is related, in large part, to uncertainties regarding: 1) the operational mechanisms that steer tumor cells to the brain, 2) the intrinsic biology of metastasis to the CNS, and 3) the foundational interactions that enable tumor cells to adapt to the brain microenvironment. Still, one of the most intriguing set of data relates to prostaglandin and matrix metalloproteinase (MMP). Although a family consists of more than 25 variants, MMP1 may have a focal role in breast cancer brain metastases.114 Not only has tumor cell-derived MMP1 been positively correlated with brain metastasis, but the protein appears to be at the center of the metastatic mayhem. MMP1 is up-regulated by prostaglandins E2 and F2α, both of which are end products of the COX2 pathway.115 Although the macro-components of the BBB include endothelial cells, astrocytes, and pericytes, the integrity of the barrier is dependent on multiple micro-proteins, two of which are Occludin and Claudin. The implication of COX2 in breaching the critical first step of the brain metastasis may be through MMP1-mediated degradation of these two junctional adhesion molecules, creating gaps in the tight confluences of the BBB.116 This belief is in accord with the finding that inhibition of COX2 suppressed not only expression of MMP1 but also transmigration of tumor cell through blood-brain barrier constructs. Consistent was also the demonstration that the inhibitory effects of COX2 knockdown could be reversed by addition of prostaglandins. Adaptation to a different microenvironment is the critical second step in the metastatic process. Abundant in the brain, astrocytes have the ability to support the propagation of metastatic tumor-initiating cells.117 Interestingly, PG-E2 and -F2α can, in a paracrine-like manner, also activate astrocytes, which may affect the generation of a functional niche compatible for tumor growth and survival. The postulated central role of the MMPs is not the absolute answer but does provide revocable links to the three uncertainties listed above. Moreover, these studies provide a glimmer of hope to address breast cancer brain metastasis.
Conclusion
Despite all the accumulated information, the mechanisms responsible for tumor resistance and brain metastases are important areas of research that are still only partially resolved. In addition, inquiry focused on receptor-targeted anti-tumor effect is still ongoing; and controversy still exists regarding the proposed mechanisms. Yet another subtlety concerns the respective significance of the other HER family members. And while HER2 overexpression occurs in non-invasive breast cancer,118 it is intriguingly possible for the receptor, in some cases, to undergo pathologic conversion even earlier and initiate the neoplastic process. In essence, ascertaining the time when HER2 becomes functionally perfidious may be especially relevant in breast cancer as early diagnosis has proven to be clinically beneficial.
Lastly, that perceptibly large knowledge gaps still exist in achieving full mastery of this formidable receptor is concordant with the quote “science is piecemeal revelation” (Oliver Wendell Holmes Sr.).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors do not have any relationship, financial or otherwise (ie, support in the form of employment, consultancies, honoraria, stock ownership and options, expert testimony, grants or patents received or pending, or royalties) with manufacturers of all agents described that influenced the writing of this manuscript.
References
1. Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981;290(5803):261–264. doi:10:1038/290261a0
2. Vennstrom B, Bishop JM. Isolation and characterization of chicken DNA homologous to the two putative oncogenes of avian erythroblastosis virus. Cell. 1982;28(1):135–143. doi:10.1016/0092-8674(82)90383-x
3. Schechter AL, Stem DF, Vaidyanathan L, et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 1984;312(5994):513–516. doi:10.1038/312513a0
4. King CR, Kraus MH, Aaronson SA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229(4717):974–976. doi:10.1126/science.2992089
5. Akiyama T, Sudo C, Ogawara H, Toyoshima K, Yamamoto T. The product of the human c-erbB-2 gene: a 185 kilodalton glycoprotein with tyrosine kinase activity. Science. 1986;232(4758):1644–1646. doi:10.1126/science.3012781
6. van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol. 1994;10:251–337. doi:10.1146/annurev.cb.10.110194.001343
7. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. doi:10.1016/j.cell.2006.05.013
8. van Lengerich B, Agnew C, Puchner EM, Huang B, Jura N. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A. 2017;114(14):E2836–E2845. doi:10.1073/pnas.1617994114
9. Jones FE, Stern DF. Expression of dominant-negative ErbB2 in the mammary gland of transgenic mice reveals a role in lobuloalveolar development and lactation. Oncogene. 1999;18(23):3481–3490. doi:10.1038/sj.onc.1202698
10. Hynes NE, Stern DF. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta. 1994;1198(2–3):165–184. doi:10.1016/0304-419x(94)90012-4
11. McCann AH, Dervan PA, O’Regan M, et al. Prognostic significance of c-erbB-2 and estrogen receptor status in human breast cancer. Cancer Res. 1991;51(12):3296–3303.
12. Menard S, Fortis S, Castiglioni F, Agresti R, Balsari A. HER2 as a prognostic factor in breast cancer. Oncology. 2001;61(Suppl 2):67–72. doi:10.1159/000055404
13. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101
14. von Minckwitz G, Du Bois A, Schmidt M, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2–positive advanced breast cancer: a German breast group 26/breast international group 03–05 study. J Clin Oncol. 2009;27(12):1999–2006. doi:10.1200/JCO.2008.19.6618
15. Burgess AW, Cho HS, Eigenbrot C, et al. An open-and- shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541–552. doi:10.1016/s1097-2765(03)00350-2
16. Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;237(4811):178–182. doi:10.1126/science.2885917
17. Pegram Md, Lipton A, Hayes DF, et al. Phase II study of receptor‐enhanced chemosensitivity using recombinant humanized anti‐p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu‐overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol. 1998;16(8):2659–2671. doi:10.1200/JCO.1998.16.8.2659
18. Baselga J, Tripathy D, Mendelsohn J, et al. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu‐overexpressing metastatic breast cancer. Semin Oncol. 1999;26(4 Suppl 12):78–83.
19. Quirke P, Pickles A, Tuzi NL, Mohamdee O, Gullick WJ. Pattern of expression of c-erbB2 oncoprotein in human fetuses. Br J Cancer. 1989;60(1):64–69. doi:10.1038/bjc.1989.221
20. Ozcelik C, Erdmann B, Pilz B, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci USA. 2002;99(13):8880–8885. doi:10.1073/pnas.122249299
21. Hong TT, Smyth JW, Gao D, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PloS Biol. 2010;8(2):e1000312. doi:10.1371/journal.pbio.1000312
22. Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16(7):1647–1655. doi:10.1093/emboj/16.7.1647
23. Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924):756–760. doi:10.1038/nature01392
24. Agus DB, Akita RW, Fox WD, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–137. doi:10.1016/s1535-6108(02)00097-1
25. Baselga J, Cortés J, Kim SB, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–119. doi:10.1056/NEJMoa1113216
26. Swain SM, Miles D, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(4):519–530. doi:10.1016/S1470-2045(19)30863-0
27. Zhang M, Zhang Z, Garmestani K, et al. Activating Fc receptors are required for antitumor efficacy of the antibodies directed toward CD25 in a murine model of adult T-cell leukemia. Cancer Res. 2004;64(16):5825–5829. doi:10.1158/0008-5472.CAN-04-1088
28. Stavenhagen JB, Gorlatov S, Tuaillon N, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 2007;67(18):8882–8890. doi:10.1158/0008-5472.CAN-07-0696
29. Rugo HS, Im SA, Cardoso F, et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer. A phase 3 randomized clinical trial. JAMA Oncol. 2021;7(4):573. doi:10.1001/jamaoncol.2020.7932
30. Tan AR, Im SA, Mattar A, et al. Subcutaneous administration of the fixed-dose combination of trastuzumab and pertuzumab in combination with chemotherapy in HER2-positive early breast cancer: primary analysis of the Phase III, multicenter, randomized, open-label, two-arm FeDeriCa study.
31. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791. doi:10.1056/NEJMoa1209124
32. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382(7):610–621. doi:10.1056/NEJMoa1914510
33. Powell CA, Modi S, Iwata H, et al. Pooled analysis of drug-related interstitial lung disease (ILD) in 8 single-arm trastuzumab deruxtecan (T-DXd) studies.
34. Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659. doi:10.1158/0008-5472
35. Xia W, Liu L-H, Ho P, Spector NL. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW72016. Oncogene. 2004;23(3):646–653. doi:10.1038/sj.onc.1207166
36. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi:10.1056/NEJMoa064320
37. Geyer CE, Forster J, Cameron MD. Lapatinib plus capecitabine in breast cancer. N Engl J Med. 2007;356(27):1471–1472. doi:10.1056/NEJMc070182
38. Chan A, Delaloge S, Holmes FA, et al.; for the ExteNET Study Group. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367–377. doi:10.1016/S1470-2045(15)00551-3
39. Saura C, Oliveira M, Feng YH, et al. Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2-positive metastatic breast cancer previously treated with >2 HER2-directed regimens: Phase III NALA Trial. J Clin Oncol. 2020;38(27):3138–3149. doi:10.1200/JCO.20.00147
40. Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–3965. doi:10.1158/0008-5472.CAN-03-2868
41. Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92(15):6981–6985. doi:10.1073/pnas.92.15.6981
42. Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003;278(3):1671–1679. doi:10.1074/jbc.M207289200
43. Ellis LM, Staley CA, Liu W, et al. Down-regulation of vascular endothelial growth factor in a human colon carcinoma cell line transfected with an antisense expression vector specific for c-src. J Biol Chem. 1998;273(2):1052–1057. doi:10.1074/jbc.273.2.1052
44. Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375(6532):577–581. doi:10.1038/375577a0
45. Sinibaldi D, Wharton W, Turkson J, Bowman T, Pledger WJ, Jove R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene. 2000;19(48):5419–5427. doi:10.1038/sj.onc.1203947
46. Mather B, Viswanathan K, Miller K, Long T. Michael addition reactions in macromolecular design for emerging technologies. Prog Polym Sci. 2006;31(5):487–531. doi:10.1016/j.progpolymsci.2006.03.001
47. Zhao XQ, Xie JD, Chen XG, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol. 2012;82(1):47–58. doi:10.1124/mol.111.076299
48. Dai CL, Tiwari AK, Wu CP, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68(19):7905–7914. doi:10.1158/0008-5472.CAN-08-0499
49. Pheneger T, Bouhana K, Anderson D, et al. In vitro and in vivo activity of ARRY-380: a potent, small molecule inhibitor of ErbB2. Cancer Res. 2009;69(9):1795.
50. Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382(7):597–609. doi:10.1056/NEJMoa1914609
51. Rimawi M, Ferrero JM, de la Haba-rodriguez J, et al. First-line trastuzumab plus an aromatase inhibitor, with or without pertuzumab, in human epidermal growth factor receptor 2-positive and hormone receptor-positive metastatic or locally advanced breast cancer (PERTAIN): a randomized, open-label phase II trial. J Clin Oncol. 2018;36(28):2826–2835. doi:10.1200/JCO.2017.76.7863]
52. Schmid P, Rugo HS, Adams S, et al.; for IMpassion130 Investigators. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44–59. doi:10.1016/S1470-2045(19)30689-8
53. Cortes J, Cescon DW, Rugo HS, et al.; for KEYNOTE-355 Investigators. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–1828. doi:10.1016/S0140-6736(20)32531-9
54. Dieci MV, Griguolo G, Miglietta F, Guarneri V. The immune system and hormone-receptor positive breast cancer: is it really a dead end? Cancer Treat Rev. 2016;46:9–19. doi:10.1016/j.ctrv.2016.03.011
55. Li S, Chen L, Jiang J. Role of programmed cell death ligand-1 expression on prognostic and overall survival of breast cancer: a systematic review and meta-analysis. Medicine. 2019;98(16):e15201. doi:10.1097/MD.0000000000015201
56. Xu J, Guo X, Jing M, Sun T. Prediction of tumor mutation burden in breast cancer based on the expression of ER, PR, HER-2, and Ki-67. Onco Ther. 2018;11:2269–2275. doi:10.2147/OTT.S159830
57. Alva AS, Mangat PK, Garrett-Mayer E, et al. Pembrolizumab in patients with metastatic breast cancer with high tumor mutational burden: results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J Clin Oncol. 2021;39(22):2443–2451. doi:10.1200/JCO.20.02923
58. Loi S, Giobbe-Hurder A, Gombos A, et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b–2 trial. Lancet Oncol. 2019;20(3):371–382. doi:10.1016/S1470-2045(18)30812-X
59. Chung A, Cui X, Audeh W, Giuliano A. Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer. 2013;13(4):223–232. doi:10.1016/j.clbc.2013.04.001
60. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. doi:10.1002/j.1460-2075.1992.tb05481.x
61. Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. doi:10.1093/intimm/8.5.765
62. Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011;28(3):682–688. doi:10.1007/s12032-010-9515-2
63. Song M, Chen D, Lu B, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS One. 2013;8(6):e65821. doi:10.1371/journal.pone.0065821
64. Tsang JY, Au WL, Lo KY, et al. PD-L1 expression and tumor infiltrating PD-1+ lymphocytes associated with outcome in HER2+ breast cancer patients. Breast Cancer Res Treat. 2017;162(1):19–30. doi:10.1007/s10549-016-4095-2
65. Baptista MZ, Sarian LO, Derchain SF, Pinto GA, Vassallo J. Prognostic significance of PD-L1 and PD-L2 in breast cancer. Hum Pathol. 2016;47(1):78–84. doi:10.1016/j.humpath.2015.09.006
66. Bae SB, Cho HD, Oh MH, et al. Expression of programmed death receptor ligand 1 with high tumor-infiltrating lymphocytes is associated with better prognosis in breast cancer. J Breast Cancer. 2016;19(3):242–251. doi:10.4048/jbc.2016.19.3.242
67. Dill EA, Gru AA, Atkins KA, et al. PD-L1 expression and Intratumoral heterogeneity across breast cancer subtypes and stages: an assessment of 245 primary and 40 metastatic tumors. Am J Surg Pathol. 2017;41(3):334–342. doi:10.1097/PAS.0000000000000780
68. Herbst RS, Soria J-C, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011
69. Kim JW, Eder JP. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology. 2014;28(Suppl 3):15–28.
70. Kim HR, Ha SJ, Hong MH, et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci Rep. 2016;6:36956. doi:10.1038/srep36956
71. de Kleijn S, Langereis JD, Leentjens J, et al. IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS One. 2013;8(8):e72249. doi:10.1371/journal.pone.0072249
72. Burstein HJ, Sun Y, Dirix LY, et al. Neratinib, an irreversible erbB receptor tyrosine kinase inhibitor, in patients with advanced erbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–1307. doi:10.1200/JCO.2009.25.8707
73. Olayioye MA, Graus-Porta D, Beerli RR, Rohrer J, Gay B, Hynes NE. ErbB-1 and erbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol Cell Biol. 1998;18(9):5042–5051. doi:10.1128/mcb.18.9.5042
74. Alimandi M, Romano A, Curia MC, et al. Cooperative signaling of erbB3 and erbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821.
75. Kokai Y, Myers JN, Wada T, et al. Synergistic interaction of p185c-neu and the EGF receptor leads to transformation of rodent fibroblasts. Cell. 1989;58(2):287–292. doi:10.1016/0092-8674(89)90843-x
76. Boulbes DR, Arold ST, Chauhan GB, et al. HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol Oncol. 2015;9(3):586–600. doi:10.1016/j.molonc.2014.10.011
77. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. doi:10.1158/2159-8290.CD-12-0349
78. Lee JW, Soung YH, Seo SH, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006;12(1):57–61. doi:10.1158/1078-0432.CCR-05-0976
79. Segatto O, King CR, Pierce JH, DiFiore PP, Aaronson SA. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol Cell Biol. 1988;8(12):5570–5574. doi:10.1128/mcb.8.12.5570
80. Liu PCC, Liu X, Li Y, et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol Ther. 2006;5(6):657–664. doi:10.4161/cbt.5.6.2708
81. Molina MA, Saez R, Ramsey EE, et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin Cancer Res. 2002;8(2):347–353.
82. Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-1 receptor signaling and resistance to trastuzumab. J Natl Cancer Inst. 2001;93(24):1852–1857. doi:10.1093/jnci/93.24.1852
83. Harris LN, You F, Schnitt SJ, et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res. 2007;13(4):1198–1207. doi:10.1158/1078-0432.CCR-06-1304
84. Nahta R, Yuan LXH, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-1 receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. doi:10.1158/0008-5472.CAN-04-3841
85. Lai A, Sarcevic B, Prall OWJ, Sutherland RL. Insulin/insulin-like growth factor-I and estrogen cooperate to stimulate cyclin E-Cdk2 activation and cell cycle progression in MCF-7 breast cancer cells through differential regulation of cyclin E and p21WAF1/Cip. J Biol Chem. 2001;276(28):25823–25833. doi:10.1074/jbc.M100925200
86. Nicholson RI, Hutcheson IR, Knowlden JM, et al. Nonendocrine pathways and endocrine resistance; observations with antiestrogens and signal transduction inhibitors in combination. Clin Cancer Res. 2004;10(1 Pt 2):346S–354S. doi:10.1158/1078-0432.ccr-031206
87. Tang CK, Perez C, Grunt T, Waibel C, Cho C, Lupu R. Involvement of heregulin-beta2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res. 1996;56(14):3350–3358.
88. Nicholson RI, Staka C, Boyns F, Hutcheson IR, Gee JM. Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endocr Relat Cancer. 2004;11(4):623–641. doi:10.1677/erc.1.00778
89. Massarweh S, Osborne CK, Jiang S, et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006;66(16):8266–8273. doi:10.1158/0008-5472.CAN-05-4045
90. Kaufman B, Mackey JR, Clemens M, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol. 2009;27(33):5529–5537. doi:10.1200/JCO.2008.20.6847
91. Johnston S, Pippen J, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538–5546. doi:10.1200/JCO.2009.23.3734
92. Wang YC, Morrison G, Gillihan R, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers – role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13(6):R121. doi:10.1186/bcr3067
93. Xia W, Bacus S, Hegde P, et al. A model of acquired auto-resistance to a potent erbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A. 2006;103(20):7795–7800. doi:10.1073/pnas.0602468103]
94. Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92(15):1210–1216. doi:10.1093/jnci/92.15.1210
95. Chandarlapaty S, Scaltriti M, Angelini P, et al. Inhibitors of HSP90 block p95-HER2 signaling in trastuzumab-resistant tumors and suppress their growth. Oncogene. 2010;29(3):325–334. doi:10.1038/onc.2009.337
96. Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–3277. doi:10.1200/JCO.2009.25.9820
97. Arvold MD, Oh KS, Niemierko A, et al. Brain metastases after breast conserving therapy and systemic therapy: incidence and characteristics by biologic subtype. Breast Cancer Res Treat. 2012;136(1):153–160. doi:10.1007/s10549-012-2243-x
98. Gupta P, Srivastava SK. HER2 mediated de novo production of TGFβ leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer. Mol Oncol. 2014;8(8):1532–1547. doi:10.1016/j.molonc.2014.06.006
99. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3(3):155–166. doi:10.1038/nrm757
100. Hohensee I, Lamszus K, Riethdorf S, et al. Frequent genetic alterations in EGFR- and HER2-driven pathways in breast cancer brain metastases. Am J Pathol. 2013;183(1):83–95. doi:10.1016/j.ajpath.2013.03.023
101. Da Silva L, Simpson PT, Smart CE, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010;12(4):R46. doi:10.1186/bcr2603
102. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi:10.1038/35052073
103. Law AJ, Shannon Weickert C, Hyde TM, Kleinman JE, Harrison PJ. Neuregulin-1 (NRG-1) mRNA and protein in the adult human brain. Neuroscience. 2004;127(1):125–136. doi:10.1016/j.neuroscience.2004.04.026
104. Gaedcke J, Traub F, Milde S, et al. Predominance of the basal type and HER-2/neu type in brain metastasis from breast cancer. Mod Pathol. 2007;20(8):864–870. doi:10.1038/modpathol.3800830
105. Wikman H, Lamszus K, Detels N, et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012;14(2):R49. doi:10.1186/bcr3150
106. Zhang L, Ridgway L, Wetzel M, et al. The identification and isolation of breast cancer CTCs with brain metastatic competence. Sci Transl Med. 2013;5(180):84–93. doi:10.1126/scitranslmed.3005109
107. Zetser A, Bashenko Y, Miao H-Q, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741.
108. Ridgway L, Wetzel MA, Epstein A, Marchetti D. Heparanase-induced GEF-H1 signaling regulates the cytoskeletal dynamics of brain metastatic breast cancer cells. Mol Cancer Res. 2013;10(6):689–702. doi:10.1158/1541-7786.MCR-11-0534
109. Choi YK, Woo SM, Cho SG, et al. Brain-metastatic triple-negative breast cancer cells regain growth ability by altering gene expression patterns. Cancer Genomics Proteomics. 2013;10(6):265–275.
110. Ueno Y, Sakurai H, Tsunoda S, et al. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int J Cancer. 2008;123(2):340–347. doi:10.1002/ijc.23465
111. Bachelot T, Romieu G, Campone M, et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 2013;14(1):64–71. doi:10.1016/S1470-2045(12)70432-1
112. Freedman RA, Gelman RS, Anders CK, et al. TBCRC 022: phase II trial of neratinib + capecitabine for patients with human epidermal growth factor receptor 2 breast cancer brain metastases. J Clin Oncol. 2019;37(13):1081–1089. doi:10.1200/JCO.18.01511
113. Morikawa A, Peereboom DM, Thorsheim HR, et al. Capecitabine and lapatinib uptake in surgically resected brain metastases from metastatic breast cancer patients: a prospective study. Neuro Oncol. 2015;17(2):289–295. doi:10.1093/neuonc/nou141
114. Poola I, DeWitty RL, Marshalleck JJ, Bhatnagar R, Abraham J, Leffall LD. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis. Nat Med. 2005;11(5):481–483. doi:10.1038/nm1243
115. Nørregaard R, Kwon TH, Frøkiær J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res Clin Pract. 2015;34(4):194–200. doi:10.1016/j.krcp.2015.10.004
116. Lee KY, Kim YJ, Yoo H, Lee SH, Park JB, Kim HJ. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res. 2011;31(12):4307–4313.
117. Xing F, Kobayashi A, Okuda H, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5(3):384–396. doi:10.1002/emmm.201201623
118. Chaisson K, Rivere A, Corsetti R, Weiss T, Fuhrman GM. A potential additional variable to consider in the surgical treatment of ductal carcinoma in situ. Ochsner J Winter. 2017;17:341–344.
119. Burstein HJ, Keshaviah A, Baron AD, et al. Trastuzumab plus vinorelbine or taxane chemotherapy for HER2-overexpressing metastatic breast cancer: the trastuzumab and vinorelbine or taxane study. Cancer. 2007;110(5):965–972. doi:10.1002/cncr.22885
120. Blackwell KL, Burstein H, Storniolo A, et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28(7):1124–1130. doi:10.1200/JCO.2008.21.4437
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.