")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Necrotizing RPGN with linear anti IgG deposits in a patient with history of granulomatosis with polyangiitis: a case report
Authors Parekh N, Epstein E, El-sayegh S
Received 30 January 2014
Accepted for publication 27 February 2014
Published 27 November 2014 Volume 2014:7 Pages 441—446
DOI https://doi.org/10.2147/IJNRD.S61621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Ninad Parekh, Edward Epstein, Suzanne El-Sayegh
Department of Medicine, Division of Nephrology, Staten Island University Hospital, Staten Island, NY, USA
Introduction: Diagnosing the etiology of a rapidly progressive glomerulonephritis is of vital importance to guide appropriate therapeutic management. This case highlights the complexity involved in establishing diagnosis when presentation is atypical. In certain cases diagnosis cannot be established based on clinical presentation or biopsy findings alone, and critical analysis of biopsy findings in context of clinical presentation is crucial to guide the clinical decision-making process.
Case presentation: A 47-year-old Hispanic male with history of granulomatosis with polyangiitis (GPA) in remission on azathioprine, presented with fatigue and lethargy. Physical examination was unremarkable. Laboratory data revealed elevated creatinine and otherwise normal electrolytes. Urinalysis showed numerous dysmorphic red blood cells with few red cell casts. His serologic results were all negative except anti-proteinase-3 antibody at very low titers. Kidney biopsy showed necrotizing crescentic glomerulonephritis with linear immunoglobulin G staining along the basement membrane.
Conclusion: This case presented conflicting serologic and histopathologic findings. The presence of anti-proteinase-3 antibody supported diagnosis of recurrence of GPA. However, linear staining of immunoglobulin G (IgG) on immunofluorescence (IF) staining of renal biopsy supported anti-glomerular basement membrane (GBM) disease. The treatment of anti-GBM disease and GPA both involve immunosuppression with prednisone and cyclophosphamide. However, patients with anti-GBM disease are also treated with plasmapheresis early in the disease presentation to prevent further damage. The patient with GPA, on the other hand, was shown to benefit from plasmapheresis only in the case of severe renal disease (serum creatinine level more than 5 mg/dL) or pulmonary hemorrhage. In this case, since the patient did not have detectable circulating anti-GBM antibody, the decision was made not to proceed with plasmapheresis. The patient was treated with a standard immunosuppressive regimen consisting of prednisone and cyclophosphamide with partial renal recovery at 2 months.
Keywords: Necrotizing RPGN, Anti-GBM disease, GPA, ANCA - associated vasculitis, dual antibody-positive disease
Introduction
Acute worsening of kidney function associated with glomerular hematuria, as evidenced by red cell casts or dysmorphic red blood cells, points towards glomerulonephritis. Glomerulonephritis causing progressive loss of renal function over a relative short period of time is called rapidly progressive glomerulonephritis (RPGN).1 Several serologic studies may help in evaluating potential cause of glomerulonephritis. However, kidney biopsy is essential to provide histologic confirmation of the diagnosis. To clinician’s surprise, coexisting pathologies can often be found on kidney biopsy. In a patient with known glomerulonephritis that has responded well to treatment and has been in remission, recurrent disease would be the most likely etiology of worsening renal function with glomerular hematuria. Nevertheless, a second glomerular disease may coexist on kidney biopsy. Therefore, it is very important to establish a diagnosis before treatment decisions are made.
Among various causes of RPGN, granulomatosis with polyangiitis (GPA) (formerly known as Wegener’s granulomatosis) and anti-glomerular basement membrane (GBM) disease (anti - GBM disease) are two disease processes very difficult to differentiate clinically due to similar presentation. Wegener’s granulomatosis was renamed as GPA during the 2012 International Chapel Hill Consensus Conference that provided revised nomenclature and definitions of vasculitides.2 GPA is a small vessel vasculitis characterized by granuloma formation and immune deposits of affected vasculature.3 GPA is part of a spectrum of diseases known as anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV), which includes GPA, microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA; formerly known as Churg–Strauss syndrome), and renal limited vasculitis.3,4
Clinically, three major organ systems are affected in patients with GPA – ear, nose and throat; respiratory system; and kidneys. However, not all patients will have all of the manifestations, and it is not uncommon for AAV to have renal-limited presentation. Classically described anti-GBM disease by Dr Goodpasture (thereby called Goodpasture’s disease) involves lungs and kidneys.5 However, glomerulonephritis caused by anti-GBM disease without pulmonary involvement is also well described.5 Therefore, establishing diagnosis by clinical presentation alone may be challenging.
Even though serologic tests are not required for diagnosis of GPA, they are very useful in clinical practice. Circulating ANCA is positive in approximately 92% of patients with active GPA,6 whereas, anti-GBM disease is characterized by antibodies directed against an antigen present in the GBM.5,6 Presence of circulating anti-GBM antibodies is crucial for diagnosis of anti-GBM disease. But the serologic tests by themselves do not establish the diagnosis. Hence, kidney biopsy findings play a crucial role in establishing the diagnosis.
The hallmark of renal biopsy findings in patients with GPA is few or no immune deposits as seen by immunofluorescence (IF) and electron microscopy.4 Therefore, renal involvement in GPA is classically described as pauci-immune crescentic glomerulonephritis (PICGN). Consequently, the finding of immune deposits on kidney biopsy points towards separate etiology. On the other hand, the pathognomonic finding of anti-GBM disease is linear staining of immunoglobulin (Ig)G (rarely IgA or IgM) along the basement membrane of glomerular capillaries on IF staining.7
The diagnostic challenge arises when clinical presentation and serologic tests point towards one disease, but the biopsy findings support an alternative diagnosis. Since the treatment of these two disease (GPA and anti-GBM) involves chemotherapeutic agents (see discussion), one needs to be very cautious in clinical decision-making. Here we present a case of conflicting serology and pathology findings.
Case presentation
A 47-year-old Hispanic male presented with lower back pain, and lethargy. The symptoms started approximately 1 week before presentation. The patient denied having any upper respiratory symptoms, rash, or joint pain. The patient also denied having any gross hematuria, foamy urine, or any other urinary symptoms. The patient’s history included GPA, diagnosed by a renal biopsy 2 years ago, diabetes mellitus, hypertension, hyperlipidemia, and diverticulosis requiring colostomy.
His medication at the time included simvastatin, lisinopril, azathioprine, and insulin. His azathioprine was reduced from 200 mg/day to 100 mg/day 6 months before presentation. He had recently lost his job and insurance, and denied smoking, alcohol use, or any substance abuse. On physical examination, he was afebrile. His blood pressure was 99/51 mmHg, pulse was 74 beats/minute, and respiration rate 18 breaths/minute, height was 165 cm, and weight was 97 kg. He was in no apparent distress. The patient’s lungs were clear, heart sounds were regular without any murmur, rub, or gallop, the abdomen was soft, and no edema or skin rash was present.
Laboratory data revealed the patient’s blood urea nitrogen (BUN) to be 73 mg/dL and serum creatinine (Scr) 5.2 mg/dL with otherwise normal electrolytes, hemoglobin 12.7 mg/dL, platelets 257,000/mm3, albumin 3.5 mg/dL, and normal liver enzymes. Two years prior to current presentation his Scr level was 1.5 mg/dl. Urinalysis showed large protein, large blood, many dysmorphic red blood cells and red blood cell casts. The urine protein to creatinine ratio was 2.03 mg/mg. Renal sonogram revealed kidney sizes of 11.3 cm and 10.6 cm on right and left kidneys, respectively, without any evidence of obstruction and an unremarkable bladder with no significant postvoid residual volume.
The patient was started on high dose pulse methylprednisolone intravenously (1 g/day for 3 days), and his azathioprine dose was increased to 200 mg/day. His serology showed normal complement titers, negative hepatitis profile, and mildly elevated rheumatoid factor at 24 IU/ml (normal <14). ANCA serology showed proteinase-3 (PR3) antibody (anti-PR3, cytoplasmic c-ANCA) reactive at low levels −4.1 (<1.0 reported as negative) and negative myeloperoxidase (MPO) antibody (anti-MPO, perinuclear p-ANCA). Anti-GBM titers were negative.
Based on clinical presentation and the patient’s past medical history, it was presumed that the most likely cause of worsening renal function was a relapse of GPA. However, very low levels of anti-PR3 antibodies, and the questionable duration of the progression of the patient’s renal disease prompted the nephrology team to proceed with a repeat kidney biopsy.
The kidney biopsy showed focal segmental necrotizing and crescentic glomerulonephritis, with weak linear staining of IgG, which was not seen on previous biopsy. Out of 19 glomeruli examined, nine showed complete or near complete global sclerosis. Crescents were seen in eleven of 19 glomeruli. The other biopsy findings included moderate tubular atrophy, moderate interstitial fibrosis with inflammation, and moderate arteriosclerosis (Figures 1 and 2). The pathologist reported findings suggestive of moderate disease activity and moderate chronicity. Although the biopsy indicated some evidence of chronic disease, there was still significant active disease that should be treated. Provided that the disease process responded to the treatment, there was a possibility of at least partial recovery of renal function.
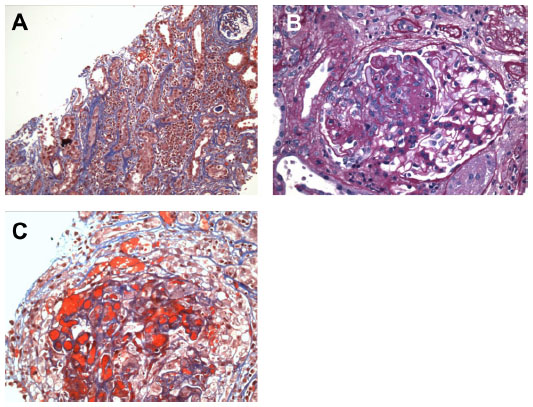 | Figure 1 Renal biopsy – light microscopy. |
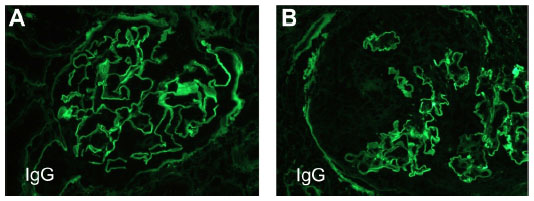 | Figure 2 Renal biopsy – immunofluorescence microscopy. |
The biopsy findings presented a treatment dilemma. The patient had a history of GPA and initially presented with PICGN and crescentic glomerulonephritis with negative IF findings 2 years previously (Figure 3). At that time, he was treated with pulse methylprednisolone followed by 6 months of oral prednisone and cyclophosphamide, which successfully induced remission. His renal function had improved; Scr decreased from 3.1 mg/dL to 1.5 mg/dL and proteinuria decreased from 2.3 g/day to <500 mg/day. He was then switched to maintenance immunosuppression with azathioprine. At the time of his initial presentation, his anti-PR3 level was >100 U/ml. After initial induction therapy, anti-PR3 level was reduced below assay limits and remained negative during follow up.
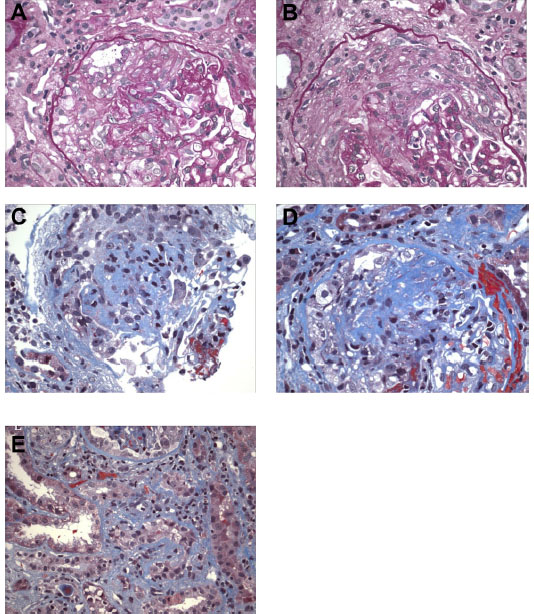 | Figure 3 Kidney biopsy from 3 years pre-presentation – light microscopy. |
This patient was treated with pulse dose of intravenous methyl prednisolone 1 g/day for 3 days followed by maintenance oral prednisone at 1 mg/kg/day. He was also treated with monthly intravenous cyclophosphamide (15 mg/kg). Three months later, the patient had partially responded with proteinuria decreasing to 800 mg/day and Cr stabilizing to 2.8 mg/dL.
Discussion
This patient presented with elevated BUN and Scr, along with hematuria and proteinuria. The presence of dysmorphic red blood cells and red blood cell casts in urine indicate that hematuria is of glomerular origin. These findings are consistent with the diagnosis of glomerulonephritis. The duration of the injury in this case is unknown, therefore a chronic glomerulonephritis as an etiology cannot be ruled out. The patient may have had disease activity of several months duration, which may have developed extensive fibrotic disease that may not be treatable with aggressive medical management. The diagnosis, therefore, requires kidney biopsy to establish the etiology, as well as to provide prognostic information regarding the potential of renal recovery with appropriate treatment. Based on clinical presentation and biopsy findings, two disease processes need to be considered in differential; GPA and anti-GBM disease.
Diagnosis of GPA is established if two or more of the following criteria are met: nasal or oral inflammation; chest radiograph demonstrating abnormalities such as nodules, fixed infiltrates, or cavities; abnormal urinary sediment showing microscopic hematuria with or without red blood cell cast; and arterial or periarteriolar granulomatous inflammation on biopsy. Diagnosis of anti-GBM disease is based on serologic and kidney biopsy findings.
Serologic tests can help differentiate between GPA and anti-GBM disease. In case of GPA, serologic tests for ANCA are positive in 92% of patients.5 The two major ANCA responsible for clinical disease and systemic vasculitis are directed against proteinase-3 (PR3) and myeloperoxidase (MPO). To detect circulating ANCA, there are two main types of assays used in clinical practices, indirect immunofluorescence (IF) assay or enzyme-linked immunosorbent assay (ELISA).6 The IF technique detects the pattern of staining of antibodies against human neutrophils, which can be described as diffuse c-ANCA pattern or p-ANCA pattern.6 C-ANCA pattern is primarily associated with PR3-ANCA and P-ANCA pattern is largely associated with MPO-ANCA. Occasionally, PR3-ANCA will demonstrate perinuclear pattern, and MPO-ANCA may be associated with cytoplasmic pattern on IF.6 The ELISAs on the other hand, are directed specifically against PR3 or MPO.8 The IF technique is more sensitive while ELISAs are more specific.8 Laboratory testing for ANCA is not standardized and therefore the diagnostic ability and reference values vary among different laboratories using different assay methods.5 The ANCA in patients with GPA is largely PR3-ANCA, but 20% of patients may have MPO-ANCA and up to 10% of patients may be ANCA negative.9
Anti-GBM antibodies can be detected by either commercially available ELISA or indirect IF assays. Indirect IF requires an experienced pathologist to perform and interpret the test, which has a very high false-negative rate, and is therefore not commercially available.10 ELISA, on the other hand, is easy to perform but may still be false-negative in patients with very low titers of anti-GBM antibody.11 Circulating anti-GBM antibodies are not detected by commercially available ELISAs in the minority of patients with anti-GBM disease. However, linear deposits of IgG along the GBM is seen by routine IF microscopy.12 Similarly, circulating ANCA is present in patients with anti-GBM disease in up to 33% of patients, with the majority being anti-MPO antibodies.13 It has also been suggested that anti-MPO and anti-PR3 antibodies may disrupt the GBM and expose the target epitopes involved in pathogenesis of anti-GBM disease.14
Histologically, renal biopsy findings in patients with GPA may range from a mild glomerulonephritis involving focal and segmental areas, to a diffuse crescentic and necrotizing glomerulonephritis. The hallmark of renal biopsy findings in patients with GPA is few or no immune deposits as seen by IF and electron microscopy.3 Therefore, renal involvement in GPA is classically described as PICGN. In anti-GBM disease, the kidney biopsy shows crescentic glomerulonephritis on light microscopy. The pathognomonic finding of anti-GBM disease is linear staining of IgG (rarely IgA or IgM) along the basement membrane of glomerular capillaries on IF staining.15 The antibodies are typically directed against the noncollagenous domain 1 (NC1) of the α-3 chain of type IV collagen.16 The antibody is typically IgG (subtypes IgG1 and IgG3) but can sometimes have IgA or IgM.16
Treatment of GPA includes immunosuppression with cyclophosphamide and steroids.17 The alternative treatment with rituximab is also equally effective.18,19 In patients with GPA presenting with severe renal dysfunction or pulmonary hemorrhage, plasmapheresis has shown to improve the outcomes, and lower the risk of end stage renal disease and death.20 Treatment of anti-GBM disease is directed against anti-GBM antibody and it consists of plasmapheresis to remove already existing antibody and other inflammatory mediators such as cytokines to reduce ongoing damage to the glomerular and pulmonary basement membranes.21 The second treatment strategy is to reduce formation of new anti-GBM antibodies by means of systemic immunosuppression, which typically comprises of prednisone and cyclophosphamide.21 It is very important to diagnose anti-GBM disease early and start treatment as soon as possible. Patients with more advanced disease presentation, with Scr > 5 mg/dL, are less likely to respond to treatment with almost all patient (approximately 92%) progressing to end-stage renal disease. Treatment with plasmapheresis is continued until titer of anti-GBM is negative. Immunosuppressive regimen typically continues for 2 to 3 months and then tapers off.22
There are case reports and case series describing patients with coexisting clinical diseases of anti-GBM antibody and AAV.23,24 In one such large case series, Levy et al found that patients with double-positive disease were significantly older compared to patients with isolated anti-GBM disease.22 Patients with double-antibody positive disease often presented with advanced renal failure, with 70% of patients requiring immediate renal replacement therapy compared with 55% of patients with isolated anti-GBM disease. Also, recovery of renal function despite appropriate immunosuppression and plasma exchange is rare. Also, recovery of renal function despite appropriate immunosuppression and plasma exchange is rare in patients with double-antibody positive disease.22 Approximately 75% of patients presenting with ANCA disease and severe renal failure requiring renal replacement therapy, tended to recover renal function with adequate treatment with immunosuppressive medications, whereas patients with anti-GBM disease are less likely to recover renal function (approximately 8%) if presenting with advanced renal failure.22 Thus, clinical progression of patients with double-antibody positive disease is worse than AAV, but similar to anti-GBM disease.
This case presented significant challenges in diagnostic and therapeutic approaches. Even though the patient had a history of GPA, and recurrence was most likely, GPA does not cause linear IgG deposition on basement membranes. The patient’s anti-PR3 level, although positive, was very low compared to his prior presentation. Also, linear IgG staining presented the possibility of the presence of anti-GBM disease. However, serologic test for the anti-GBM antibody using ELISA testing was negative. To our knowledge, this finding has not been reported in literature.
It is unclear whether there is any direct correlation between ANCA and anti-GBM antibodies. However, retrospective studies have demonstrated that in some cases, development of ANCA antibodies precede development of anti-GBM antibodies, years to decades before the onset of clinical disease.25 The anti-GBM antibody was demonstrated at low-level titers in these patients for years with rapidly rising titers shortly before development of clinical disease.25 The mechanism by which ANCA may lead to anti-GBM disease is unclear but may be associated with ANCA induced damage to the GBM.25
The treatment decision in this patient had to be based on clinical presentation, which was consistent with GPA given the prior history and positive anti-PR3 antibody. Since the patient did not present with any pulmonary hemorrhage and his renal disease was not very severe (serum Cr level <5 mg/dL), this patient would not likely benefit from plasmapheresis for treatment of GPA. If we were to consider treating the anti-GBM disease based on his renal histopathology, the treatment with plasmapheresis may be indicated. However, the duration of plasmapheresis, as well as treatment response in the case of anti-GBM is followed by anti-GBM antibody titer. In our case the titer was negative, as detected by available commercial assay. Therefore, it would have been difficult to follow the treatment response and determine the duration of plasmapheresis. Therefore, the decision was made to proceed with immunosuppression with prednisone and cyclophosphamide and not to proceed with plasmapheresis. He had partial response with Scr stabilized at 2.8 mg/dL and proteinuria decreased to 800 mg/day, 3 months later.
Conclusion
The diagnosis of glomerulonephritis is not always straightforward and requires careful interpretation of clinical, serologic, and pathologic findings. The treatment of glomerulonephritis often includes immunosuppression and may even require plasmapheresis. Clinical decision-making therefore should be made based on risk-benefit analysis of potential treatment options. This case presents a clinical dilemma challenged with conflicting serologic and histopathologic findings.
Disclosure
The authors report no conflicts of interest in this work
References
Courser WG. Rapidly progressive glomerulonephritis: classification, pathogenetic mechanisms, and therapy. Am J Kid Dis. 1988;11:449. | |
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. | |
Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33:1101–1107. | |
Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192. | |
Pusey C. Anti-glomerular basement membrane disease. Kidney Int. 2003;64:1530–1550. | |
Finkielman JD, Lee AS, Hummel AM, et al. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med. 2007;120(643):e9–e14. | |
Kluth DC, Rees AJ. Anti-glomerular basement membrane disease. J Am Soc Nephrol. 1999;10:2446–2453. | |
Niles JL, Pan GL, Collins AB, et al. Antigen-specific radioimmunoassays for anti-neutrophil cytoplasmic antibodies in the diagnosis of rapidly progressive glomerulonephritis. J Am Soc Nephrol. 1991;2:27–36. | |
Csernok E, Holle J, Hellmich B, et al. Evaluation of capture ELISA for detection of antineutrophil cytoplasmic antibodies directed against proteinase 3 in Wegener’s granulomatosis: first results from a multicentre study. Rheumatology. 2004;43:174–180. | |
Hagen EC, Daha MR, Hermans J, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int. 1998;53:743–753. | |
Salama AD, Dougan T, Levy JB, et al. Goodpasture’s disease in the absence of circulating anti-glomerular basement membrane antibodies as detected by standard techniques. Am J Kidney Dis. 2002;39:1162–1167. | |
Jia XY, Qu Z, Cui Z, Yang R, Zhao J, Zhao MH. Circulating anti-GBM autoantibodies against alpha3(IV)NC1 undetectable by commercial available enzyme-linked immunosorbent assays. Nephrology (Carlton). 2012;17:160–166. | |
Yang R, Hellmark T, Zhao J, et al. Antigen and epitope specificity of anti-glomerular basement membrane antibodies in patients with goodpasture disease with or without antineutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2007;18:1338–1343. | |
Rutgers A, Slot M, van Paassen P, van Breda Vriesman P, Heeringa P, Tervaert JW. Coexistence of anti-glomerular basement membrane antibodies and myeloperoxidase-ANCAs in crescentic glomerulonephritis. Am J Kidney Dis. 2005;46:253–262. | |
Kluth DC, Rees AJ. Anti-glomerular basement membrane disease. J Am Soc Nephrol. 1999;10:2446–2453. | |
Kalluri R, Wilson CB, Weber M, et al. Identification of the alpha 3 chain of type IV collagen as the common autoantigen in antibasement membrane disease and Goodpasture syndrome. J Am Soc Nephrol. 1995;6:1178–1185. | |
de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670–680. | |
Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–220. | |
Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232. | |
Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180–2188. | |
Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001;134:1033–1042. | |
Levy JB, Hammad T, Coulthart A, Dougan T, Pusey CD. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int. 2004;66:1535–1540. | |
Short AK, Esnault VL, Lockwood CM. Anti-neutrophil cytoplasm antibodies and anti-glomerular basement membrane antibodies: two coexisting distinct autoreactivities detectable in patients with rapidly progressive glomerulonephritis. Am J Kidney Dis. 1995;26:439–445. | |
Bonsib SM, Goeken JA, Kemp JD, Chandran P, Shadur C, Wilson L. Coexistent anti-neutrophil cytoplasmic antibody and antiglomerular basement membrane antibody associated disease – report of six cases. Mod Pathol. 1993;6:526–530. | |
Olson SW, Arbogast CB, Baker TP, OwshalimpurD, Oliver DK, Abbott KC, Yuan CM. Asymptomatic autoantibodies associate with future anti-glomerular basement membrane disease. J Am Soc Nephrol. 2011; 22:1946-1952. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.