")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanopharmaceuticals (part 1): products on the market
Authors Weissig V, Pettinger T, Murdock N
Received 23 May 2014
Accepted for publication 19 July 2014
Published 15 September 2014 Volume 2014:9(1) Pages 4357—4373
DOI https://doi.org/10.2147/IJN.S46900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Volkmar Weissig,1 Tracy K Pettinger,2 Nicole Murdock3
1Department of Pharmaceutical Sciences, Midwestern University College of Pharmacy Glendale, Glendale, AZ, USA; 2Department of Pharmacy Practice and Administrative Sciences, College of Pharmacy, Idaho State University, Pocatello, ID, USA; 3Department of Pharmacy Practice, Midwestern University College of Pharmacy Glendale, Glendale, AZ, USA
Abstract: In 2000, the National Institute of Health launched the National Nanotechnology Initiative to support, coordinate, and advance research and development of nanoscale projects. The impact of this new program on health-science related research and development became quickly visible. Broad governmental financial support advanced the start of new, and the deepening of already existing, interdisciplinary research. The anticipated merger of nanoscience with medicine quickly instigated the conceptualization of nanomedicine. The adoption of nanoscience terminology by pharmaceutical scientists resulted in the advent of nanopharmaceuticals. The term “nano” became tantamount to “cutting-edge” and was quickly embraced by the pharmaceutical science community. Colloidal drug delivery systems reemerged as nanodrug delivery systems; colloidal gold became a suspension of nano gold particles. In this review, we first review nanoscience related definitions applied to pharmaceuticals, we then discuss all 43 currently approved drug formulations which are publicized as nanopharmaceuticals, and finally we analyze clinical aspects of selected drug formulations.
Keywords: amphotericin-B, fenofibrate, nanodrugs, nanomedicine, nanoparticle, drug delivery
Introduction
Based on major advances in nanoscale material science, the National Institute of Health (NIH) started in the year 2000 the National Nanotechnology Initiative (NNI) as a federal government program in order to promote nanoscience-related research and development. The federal launch of a broad program generously supporting and coordinating the design, study, and exploration of nanomaterial has had a quick impact on health-science related research and development. Extensive governmental financial support greatly stimulated the launch of interdisciplinary research. The new concept of nanomedicine arose from merging nanoscience and nanotechnology with medicine. Pharmaceutical scientists quickly adopted nanoscience terminology, thus “creating” “nanopharmaceuticals”. Moreover, just using the term “nano” intuitively implied state-of-the-art research and became very fashionable within the pharmaceutical science community. Colloidal systems reemerged as nanosystems. Colloidal gold, a traditional alchemical preparation, was turned into a suspension of gold nanoparticles, and colloidal drug-delivery systems1 became nanodrug delivery systems.2
The exploration of colloidal systems, ie, systems containing nanometer sized components, for biomedical research was, however, launched already more than 50 years ago3–5 and efforts to explore colloidal (nano) particles for drug delivery date back about 40 years.6 For example, efforts to reduce the cardiotoxicity of anthracyclines via encapsulation into nanosized phospholipid vesicles (liposomes) began at the end of the 1970s.7,8 During the 1980s, three liposome-dedicated US start-up companies (Vestar in Pasadena, CA, USA, The Liposome Company in Princeton, NJ, USA, and Liposome Technology Inc., in Menlo Park, CA, USA) were competing with each other in developing three different liposomal anthracycline formulations. Liposome technology research culminated in 1995 in the US Food and Drug Administration (FDA) approval of Doxil®, “the first FDA-approved nanodrug”.9 Notwithstanding, it should be noted that in the liposome literature the term “nano” was essentially absent until the year 2000.
A comprehensive analysis of the worldwide state of investigational and approved nanomedicine products as of January 201210 has identified 67 commercialized nanodevices and 33 marketed nanotherapeutics. A total of 25 devices and 122 therapeutics currently in development accounted for 789 ongoing clinical trials. Our review will focus on commercialized nanotherapeutics or nanopharmaceuticals only, all of which are listed in Table 1. Nanomaterials as components of medical devices or for regenerative medicine, nanoparticles with antibacterial activities when used as surface coating for medical devices, and nanodevices used for biomarker detection like nanobiochips, nanoelectrodes, or nanobiosensors will not be included in this review.
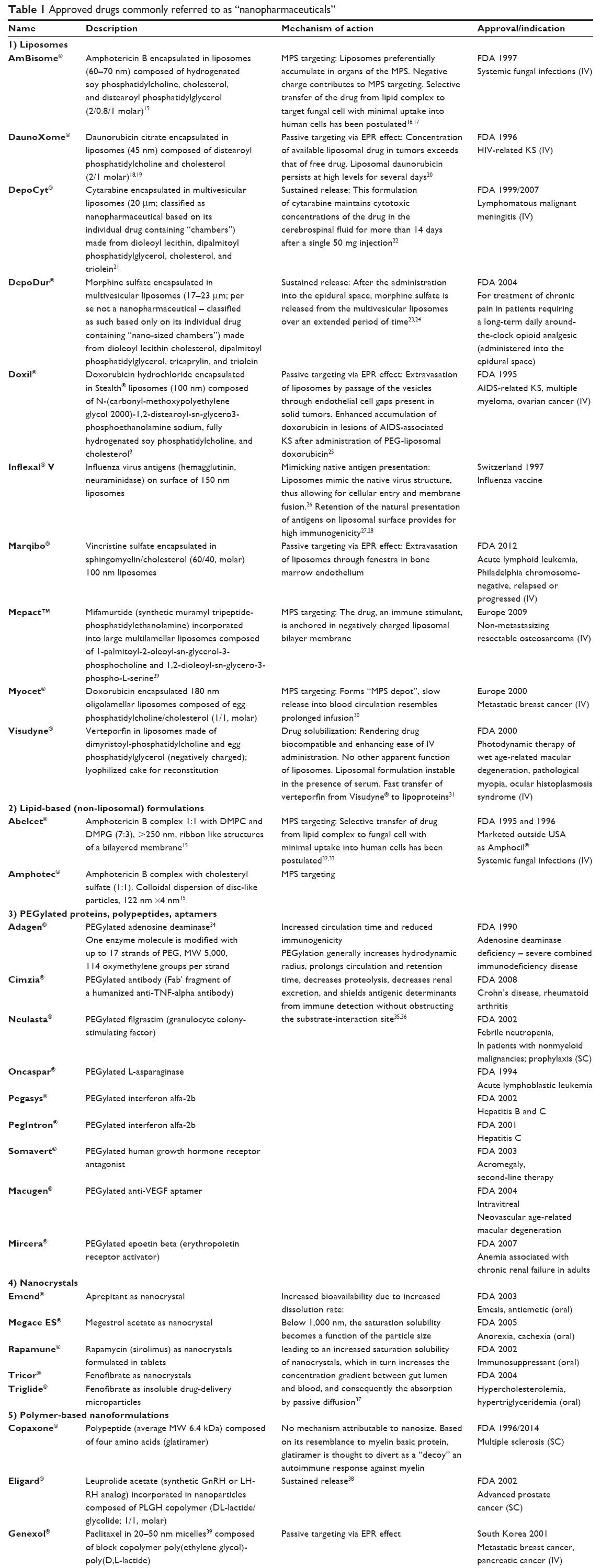 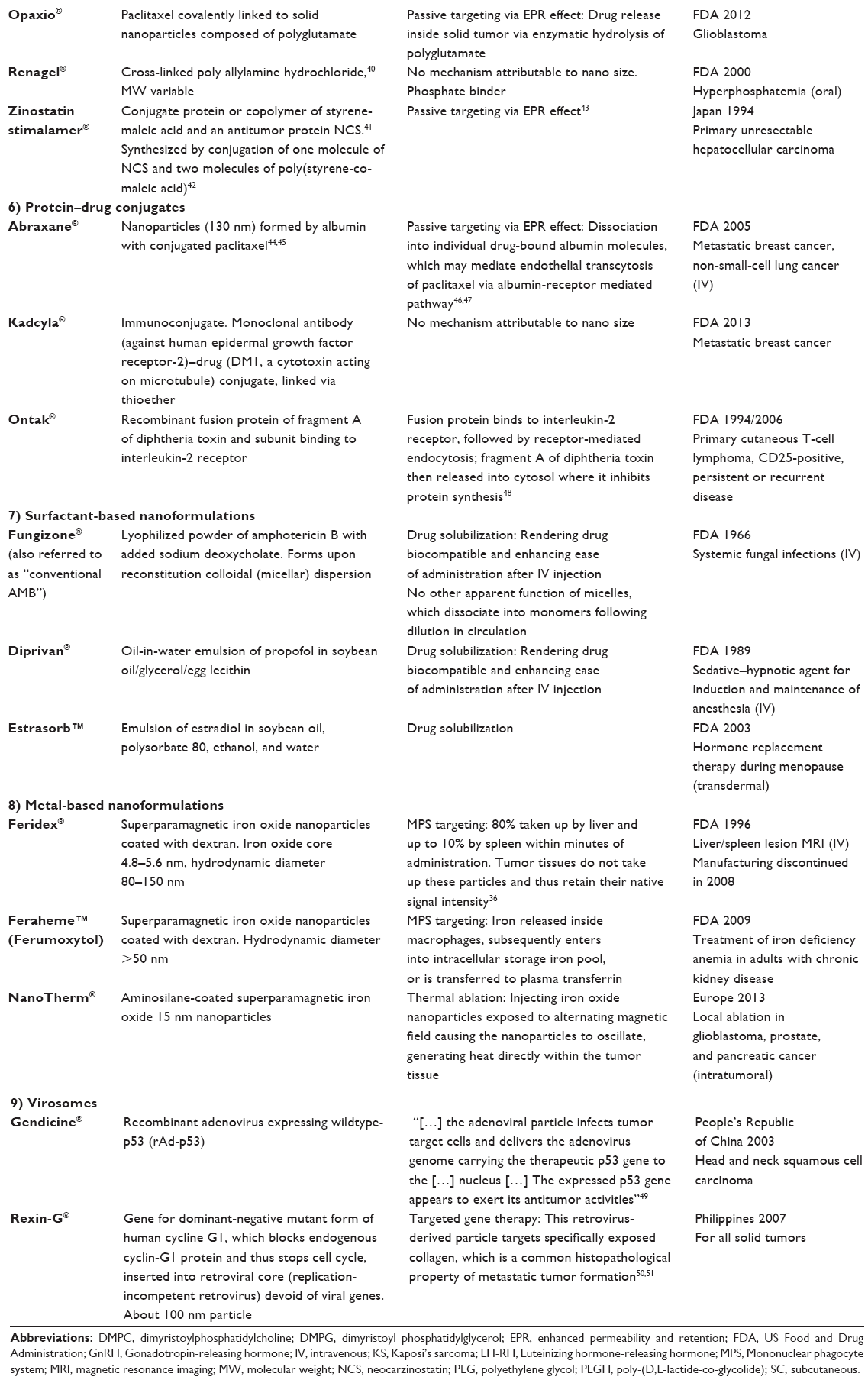 | Table 1 Approved drugs commonly referred to as “nanopharmaceuticals” |
In listing the 43 products in Table 1 as nanopharmaceuticals we followed the currently widely used custom of classifying drug products as nanopharmaceuticals mainly based on the apparent size. Each of these 43 listed drug formulations have been publicized and referred to in at least one recent peer-reviewed publication or press release/media report as “nanopharmaceutical”, “nanodrug”, or “nanomedicine”. Out of these 43 approved nanopharmaceuticals, 15 received FDA (or related foreign agency) approval before the year 2000. Further, assuming a preclinical and clinical development time of at least 10 years before any new drug formulation gets marketing approval, it becomes apparent that research and development for another 22 drugs listed in Table 1 had begun long before the NNI was launched in 2000. Only four products have been approved after the year 2010. Subsequently, attributing the successful development of the vast majority of the products listed in Table 1 to the widely advertised and NNI-supported promotion of nanoscience and nanotechnology appears questionable. We believe that the undoubted promise of nanoscience and nanoengineering for the development of unique and highly efficient therapeutics has still to materialize. We will address future developments which are based on the merger of nanotechnology and material science with pharmaceutical research in part 2 of our review paper.
What are nanopharmaceuticals?
In 2000, the NIH defined in its NNI nanotechnology as
The understanding and control of matter at dimensions between approximately 1 and 100 nanometers (nm), where unique phenomena enable novel applications not feasible when working with bulk materials or even with single atoms or molecules.11
The emphasis in this definition lies in our interpretation on the unique phenomena directly associated with this particular size range. A suitable example would be the surface plasmon resonance which is based on restricting the collective electron oscillations in a metal by limiting the shape and size of that metal. The localized surface plasmon resonance12 depends on particle size being within an extremely narrow size distribution. This causes, for example, gold nanoparticles of different sizes to appear in different colors, which neither atomic gold nor bulk gold possess. In general terms, distinctive physical, chemical, and biological properties can emerge in nanosized materials. A “true” nanomaterial therefore possesses properties which neither the bulk material nor the atoms or molecules of that same material display. In extension, engineered nanomaterials of a nearly infinite variety of sizes and shapes produced from almost any chemical substance have the potential of exhibiting unique optical, electrical, and magnetic properties.13 Logically, therefore, nanopharmaceuticals have been defined by Rivera et al as
Pharmaceuticals engineered on the nanoscale, ie, pharmaceuticals where the nanomaterial plays the pivotal therapeutic role or adds additional functionality to the previous compound.14
In conclusion, in order to be classified as a “nanopharmaceutical”, we suggest the drug product has to meet two major criteria. First, nanoengineering has to play a major role in the manufacturing process. Second, the nanomaterial used has to be either essential for the therapeutic activity or has to confer additional and unique properties to the active drug entity.
Consequently, in the following we shall apply these criteria to all drug formulations listed in Table 1. Basically, we will try to answer the question as to whether these 43 commercialized drug products are the result of nanoengineering and, further, whether the nanomaterial used is either essential for the therapeutic activity or whether it adds new functionality to the original drug molecule.
Evaluation of approved drugs as nanopharmaceuticals
Liposomes as nanopharmaceuticals
Liposomes are formed from phospholipids and cholesterol in aqueous medium; they are characterized by an aqueous inner space, or several of them, which are surrounded by one or more phospholipid bilayers. Based on the particular liposome preparation method, the size distribution of liposomes can range between 25 and 1,000 nm. Typically and most commonly used, liposome preparations display a size between 50 and 200 nm. Liposomes are used as drug carriers or drug-delivery systems based on their ability to encapsulate hydrophilic molecules in their aqueous inner space as well as hydrophobic molecules in their phospholipid bilayer membranes. Are all drugs associated with liposomes “nanopharmaceuticals”? Liposomes form spontaneously upon hydration of dry phospholipids. The self-assembly of phospholipids into bilayer membranes may take days or weeks when the water/phospholipid system is left undisturbed. Adding kinetic energy in the form of sonic or thermal energy, on the other hand, significantly accelerates the self-assembly of phospholipids into vesicles. For making liposomes suitable for therapeutic applications, their size distribution has to be controlled, which can easily be done by passing them repeatedly under elevated pressure through membranes with defined pore size. Further, a major feature of the first approved liposomal drug, Doxil®, is the presence of polyethylene glycol (PEG) chains on the liposomal surface. PEG is attached to phospholipids in the liposomal membrane via a simple one-step chemical conjugation reaction, which hardly qualifies as nanoengineering. In fact, neither probe sonication nor pressure filtration would qualify either. It should be noted, however, that new and emerging technologies like microfluidics and nanofluidics when applied to liposome preparations will potentially change this situation.
However, liposomes (when considered as nanomaterial) most certainly meet the second part of the above definition. The ability of phospholipid vesicles to incorporate an unlimited variety of compounds is used to add new functionality to low-molecular and already FDA approved drug molecules, something Gregoriadis described already back in the early 1970s as putting “old drugs in[to] new clothing”.52,53 As can be seen from Table 1, liposomal drugs can follow one of three mechanisms of action (or combinations thereof), all of which involve altering the pharmacokinetics (PK) of the free drug. First, the enhanced permeability and retention effect (EPR effect) is based on defective vascular architecture and impaired lymphatic drainage of solid tumors.54 Provided a particulate nanocarrier possesses a sufficiently long circulation half-life, a small portion will passively accumulate in the interstitial tissue of a solid tumor, while at the same time avoiding other tissues due to its inability to extravasate from healthy vasculature.55–57 The cutoff size of the gaps in the endothelial cell lining of human colon adenocarcinoma LS174T transplanted in dorsal skin chambers in severe combined immunodeficient mice, for example, was observed to be between 400 and 600 nm in diameter.58 The major strategy towards increasing the longevity of liposomes in the circulatory system is based on modifying the liposomal surface with PEG, a technology which made the development of Doxil® possible.9,57
Second, targeting of the mononuclear phagocytic system (MPS) is based on what was considered in the 1970s and 1980s a major obstacle for the utilization of liposomes as drug carriers: namely, their rapid uptake by MPS cells such as macrophages and monocytes.59 Phagocytosis has even become known as the “natural fate” of liposomes.60 Whether intravenously injected liposomes are seeking or avoiding cells and organs of the MPS system almost solely depends on the absence or presence of PEG chains on the liposomal surface.61
Third, multilamellar liposomes (MLVs) are the liposomes of choice when using them as a slow or sustained release drug carrier. MLVs are composed of multiple bilayer membranes (like an onion) with each membrane surrounding one aqueous compartment suitable for accommodating low-molecular-weight hydrophilic molecules. Once drug loaded MLVs are distributed, for example, via local injection to a specific tissue, the liposomes will slowly disintegrate via a variety of mechanisms (ideally membrane by membrane), thereby gradually releasing the encapsulated drug.
From the above it becomes obvious that the major advantages of using liposomes (altered PK, improved bioavailability, and reduced toxicity) can well be materialized in size ranges larger than 100 nm. Therefore, the NNI’s definition of nanotechnology when applied to drugs seems to “pigeonhole nanotechnology into dimensions of roughly 1 to 100 nm”62 and should be extended. This would make nanotechnology applied to wet science also more consistent with classic science, according to which colloidal solutions (in contrast to true solution or suspensions) contain particles that have at least one dimension between approximately 1 nm and 1 μm63 (by which we do not intend to suggest stretching the upper limit in the size range definition of nanotechnology to 1 μm).
Lipid-based (non-liposome) nanoformulations
The above discussion of liposomes as nanopharmaceuticals applies equally to Abelcet® and Amphotec®, though electron microscopic images or other relevant data supporting the claimed unique ribbon and disk-like structures of both formulations appear to be unavailable. Interestingly, however, the half-life and the volume of distribution of amphotericin B (AMB) administered as Amphotec® seems almost identical to that of the free drug,64 suggesting that Amphotec® quickly disintegrates upon intravenous (IV) injection.
PEGylated proteins and polypeptides as nanopharmaceuticals
Generally, therapeutically used physiological macromolecules like proteins, enzymes, and polypeptides would already qualify as nanopharmaceuticals due to their size alone. Nonetheless, nanoengineering (provided one accepts a one-step chemical reaction as such) confers additional functions or properties to the original macromolecule. PEGylation of biologically active macromolecules generally increases their hydrodynamic radius, prolongs their circulation and retention time, decreases their proteolysis, decreases their renal excretion, and shields antigenic determinants from immune detection without obstructing the substrate-interaction site.35,36 Nevertheless, attaching PEG chains to proteins as a strategy for extending their blood life and reducing their immunogenicity was developed already in the early 1970s and patented in 1979.35,65 Consequently, Enzon Pharmaceuticals (Piscataway, NJ, USA) was founded in 1981, a successful biotech company which brought a large variety of PEGylated protein pharmaceuticals to the market.
Polymer-based nanoformulations as nanopharmaceuticals
Polymer-based nanoformulations comprise a very heterogeneous group of nanosized therapeutics. Eligard®, Genexol®, Opaxio®, and Zinostatin Stimalamer® would qualify as nanopharmaceuticals when evaluated by the same criteria applied to liposomes. The therapeutically active entities are incorporated in polymer-based nanosized formulations, the result of which is significantly altered PK. Passive targeting via the EPR effect and sustained release are the subsequent mechanism of actions for these formulations. Copaxone® (a polypeptide composed of four amino acids) and Renagel® (cross-linked polyallylamine hydrochloride), on the other hand, hardly meet the above discussed criteria for a true nanopharmaceutical. First, the properties of the polymer certainly differ from that of the monomer, but what should be considered in these cases as the bulk material? Again, the uniqueness of the nano concept, as laid out by the NNI, is associated with unique phenomena a certain nanomaterial is supposed to have – unique properties which are neither displayed by the bulk material nor the individual atoms or molecules of the same material. Second, categorizing or renaming chemical polymerization reactions, as introduced by Staudinger in the early 1920s, as “nanoengineering” might appear from a chemist’s point of view as an extreme extension of definitions originally applied to “nano” and to “engineering”, and subsequently to “nanoengineering”. Third, in the case of Copaxone® and Renagel®, it remains unclear which nanomaterial is adding which additional functionality to which original drug molecule. Both drug products are efficient therapeutics made of simple polymers with a size of at least one of their dimension on the nanometer scale. Therefore, classifying them as “nanopharmaceuticals” we believe is disputable.
Protein–drug conjugates as nanopharmaceuticals
Over the last decade, albumin has gained significant attention as a potential carrier for therapeutic agents suitable for improving the pharmacokinetic profile of the free drug, and an increasing number of albumin-based therapeutics are currently in clinical trials.66 The prototype of albumin-based pharmaceuticals is undoubtedly Abraxane®, which comprises 130 nm-sized nanoparticles prepared from albumin with conjugated paclitaxel. Just like any other particulate nanocarrier, albumin particles alter the PK of the free drug, subsequently leading to its passive accumulation at the site of solid tumors via the EPR effect. In addition, following the dissociation of the protein nanoparticle into individual drug-bearing albumin molecules, specific albumin-receptor mediated cellular uptake mechanisms have been discussed.46,47
Kadcyla® and Ontak®, an immunoconjugate and a recombinant fusion protein, respectively, are prototypes for targeted therapeutics. While the former selectively delivers the drug to epidermal growth factor receptor-2-expressing cells, the latter targets cells expressing the interleukin-2 receptor. The discussed definition of nanopharmaceuticals as drugs (in the section “What are nanopharmaceuticals?”) in which the nanomaterial plays a pivotal role by adding new functions to the previous compound seems to apply to both drugs provided one accepts individual proteins as “nanomaterials”.
Surfactant-based nanoformulations
Fungizone® is a preparation consisting of a dry powdery mixture of water-insoluble AMB and sodium deoxycholate. Upon adding buffer, the deoxycholate solubilizes the drug by forming polydisperse micelles.67 Though the size of micelles lies in the nanometer range, for the consideration of micellar drug formulation as nanopharmaceuticals their stability in vivo should be considered. The critical micellar concentration (CMC) of deoxycholate is 2–6 mM (water, 20°C–25°C). Once Fungizone® has been injected into the blood stream (41 mg into 5 L blood), the deoxycholate concentration is diluted to about 0.02 mM, ie, two orders of magnitude below the CMC. Though the CMC of deoxycholate measured in blood might differ from the CMC in water, it appears as reasonable to assume that deoxycholate micelles most-likely will disintegrate upon IV injection. Subsequently, the micellar nanoformulation of AMB has no impact on the PK of the drug at all; the only function of deoxycholate is to make an IV injection possible by solubilizing the drug. Therefore, in our clinical analysis of AMB formulations below, we will compare Fungizone® as “conventional” AMB formulation with Abelcet®, Amphotec®, and AmBisome®, all of which utilize advances in lipid-based drug delivery technology.
The excipients used for Diprivan® and Estrasorb® (soybean oil, glycerol, and egg lecithin and polysorbate, ethanol, and soybean oil, respectively) are not known to form stable nanosized aggregate systems in the respective combinations used. Not surprisingly, Diprivan® has been called “liposome”,62 called “micellar preparation”,68 and named “emulsion” in the package insert provided by Gensia Sicor Pharmaceuticals, Inc (Irvine, CA, USA). Likewise, Estrasorb® is marketed as “micro-encapsulated estradiol”69 and described as estradiol “encapsulated using a micellar nanoparticle technology” in the package insert provided by Novavax, Inc (Gaithersburg, MD, USA).
Nanocrystals
Nanocrystals constitute a unique group of pharmaceuticals, as they are composed of 100% water-insoluble drug without any added excipient or any associated nanocarrier system. The dissolution of solid particles in aqueous medium is described by the Noyes–Whitney equation, according to which an increased surface area increases dissolution velocity. Therefore, micronization is widely used as a common formulation method for sparingly soluble compounds. However, on the nanometer scale another factor comes into play. The saturation solubility is a constant and depends on the chemical nature of the solid material, the dissolution medium, and the temperature. Yet below a critical size, the saturation solubility becomes also a function of the particle size. Below 1,000 nm it increases with decreasing particle size. Subsequently, drug nanocrystals possess increased saturation solubility, which in turn increases the concentration gradient between gut lumen and blood, and thereby increases the absorption by passive diffusion.37 In conclusion, nanocrystals are characterized by a unique phenomenon, ie, by an increased dissolution pressure, which can directly be attributed to their nanometer size. To what extent, or whether at all, this unique phenomenon translates into improved clinical efficacy will be discussed in the section “Fenofibrate formulation” for fenofibrate nanocrystal formulations.
Virosomes
Viruses have evolved over millions of years, perfecting their ability to insert their genetic information into mammalian host cells. Attempts to utilize that unique nature-designed ability of infecting cells for gene therapy date back to the early 1980s.70–73 Following a 10-year moratorium during which regulatory and oversight issues were addressed by governmental agencies, NIH approved the first gene transfer into humans in 1990.74 As of January 2014, worldwide, a total of about 1,400 clinical gene-therapy trials using viral vectors have received regulatory approval.75
The world’s first commercial gene therapies were approved in the People’s Republic of China and the Philippines. In October 2003, Shenzhen SiBiono GenTech (Shenzhen, People’s Republic of China) obtained a drug license from the State Food and Drug Administration of China (SFDA, Beijing, People’s Republic of China) for Gendicine®, a recombinant Ad-p53 gene therapy for head and neck squamous cell carcinoma.49,76 It should be noted, however, that the approval of Gendicine® by Chinese authorities has raised concerns among the Western medical community, mainly about genuine medical–scientific progress yielding to financial interests.77 But any discussion thereof is beyond the scope of this review.
Rexin-G®, being described as the “first targeted injectable molecular genetic medicine”,50 received its approval in the Philippines in 2007. In the United States, six clinical trials (Phase I and II) have been conducted with Rexin-G® between 2007 and 2012: four have been completed, two have been terminated. Results from these six trials have not been made available.50 In Rexin-G, a von Willebrand factor-derived collagen-binding motif was incorporated via molecular engineering into the murine leukemia virus ecotropic envelope protein while maintaining its wild-type amphotropic infectivity.78 At the same time, the retroviral core was depleted of viral genes, which were replaced by molecular engineering with a gene for a dominant-negative mutant form of human cyclin G1, which in turn is able to block the natural cell cycle. Rexin-G® embodies a nanopharmaceutical in which the nanomaterial, the original virus, was prefabricated by nature. Man-made nanomaterials created by the merger of nanotechnology and material science form the basis for Feridex®, Feraheme™, and NanoTherm®, which will be discussed next.
Metal-based nanoparticles
Feridex® comprises an aqueous colloid solution of superparamagnetic iron oxide particles (SPION) with a diameter of around 5 nm, which have been surface-modified with dextran causing an increase of the particles’ hydrodynamic diameter to up to 150 nm. Coated iron oxide nanoparticles are referred to as SPION if the overall hydrodynamic size is greater than 50 nm, while particles below 50 nm are named ultra-small SPION.79 Feridex® was FDA approved in 1996 for IV administration as a magnetic resonance imaging (MRI) contrast media.80 Cells and organs of the MPS system rapidly internalize these iron oxide particles as nonphysiological particular matter while malignant transformed cells have only a very limited ability to do so. Subsequently, using MRI, liver and spleen lesions become better distinguishable from nontransformed surrounding tissues. The production of Feridex® was discontinued in 2008.
Ferumoxytol (Feraheme™) has been FDA approved for the treatment of iron deficiency anemia in adult patients with chronic kidney disease.81 Ferumoxytol releases iron inside macrophages of the MPS system. Iron then enters into either the intracellular storage iron pool or is transferred to plasma transferrin. Ferumoxytol has also been discussed as a new superparamagnetic iron oxide colloidal blood pool contrast agent82 and is currently under development as a novel imaging agent for MRI-based diagnosis of cancer and cardiovascular diseases (see Part 2 of this review).
NanoTherm®, developed by MagForce Ag (Germany), and most recently approved in Europe, is a therapeutic product which perhaps meets most closely the above-discussed criteria for a nanopharmaceutical. NanoTherm® has been engineered from nonphysiological inorganic material on the nanoscale with the nanomaterial playing the pivotal therapeutic role. NanoTherm® represents an example of inorganic systems currently under development which:
Open novel horizons for diagnosis, imaging and therapy mainly because of their nanometer-size and their high surface area to volume ratios which allow for specific functions that are not possible in the micrometer-size particles.14
NanoTherm® comprises aminosilane-coated 15 nm-sized SPION, which are directly introduced into the solid tumor mass. Exposure to a magnetic field that changes its polarity 100,000 times per second causes these particles to significantly increase their core temperature. Depending on the length of exposure to the oscillating magnetic field, the achievable intratumoral temperatures vary and either directly destroy tumor cells (thermal ablation) or sensitize them for chemotherapy (hyperthermia). Since most of the nanoparticles stay at the treatment area, follow-up treatments are possible.
Clinical data
In this section we will review the clinical benefits (efficacy, safety, tolerability, adverse drug reactions [ADRs], and cost-related aspects) of conventional versus new nanoformulations of two therapeutic agents: AMB and fenofibrate. According to our assessment, the new nanoformulations offer improved clinical benefit as compared to the conventional product in the case of AMB, but not for fenofibrate.
AMB formulations
Overview
Conventional AMB, Fungizone® AMB deoxycholate, has been available for use and considered the “gold standard” of therapy to treat invasive systemic fungal infections since the early 1960s.83,84 Despite broad-spectrum activity, the successful clinical use of Fungizone® is limited by accompanying high rates of nephrotoxicity. This adverse effect becomes particularly important when used in patients for long periods of time (cumulative dose), if there is impaired renal function at baseline, and/or if it is used concomitantly with other nephrotoxic agents. In fact, rates of renal dysfunction have been reported to be as high as 53% with conventional AMB.85
With conventional AMB use, clinicians have been forced to alter the dose, frequency, and/or duration in patients who develop acute kidney injury (AKI) while being treated. Such changes, while avoiding irreversible damage to the kidney, may make therapy subtherapeutic. In fact, higher mortality rates have been reported in patients who developed AKI compared to those who retained normal kidney function while receiving conventional AMB.86 Therefore, the development of agents which allow effective dosing may have a dramatic effect on clinical cure and mortality rates. The high rates of nephrotoxicity combined with infusion-related reactions (fevers, rigors, chills) that patients experience early in therapy, has earned conventional AMB its nickname of “amphoterrible”. During the 1980s and early 1990s, new formulations based on advances in liposome- and lipid-based drug-delivery technology were developed in efforts to improve successful AMB use. The main goal was to allow higher doses over a prolonged period of time with decreased nephrotoxicity. Between 1995 and 1997, the FDA approved three lipid-based AMB formulations – Abelcet®, Amphotec®, and AmBisome® (for brief description see Table 1) – which were labeled “nanoformulations” following the launch of the NNI in 2000. These new nanoformulations of AMB seem to take advantage of higher rates of tissue penetration in the liver, spleen, pulmonary tissue, and brain in a dose-dependent fashion, while renal AMB levels are comparable to those obtained with conventional AMB.87 This effect may allow higher levels of nanoformulated AMB to penetrate infected areas while maintaining or decreasing the exposure to the kidney. AmBisome®, the smallest (diameter of 60–70 nm) of the three nanoformulations, has been reported to have four- to seven-times higher concentrations in the brain tissue than any of the other formulations, while Abelcet® appears to have superior pulmonary perfusion compared to the other agents.88 Clinical trials have yet to be conducted to determine whether there are efficacy differences between the three nanoformulations dependent on the site of the infection.
Mortality
Several prospective studies have established noninferiority of the nanoformulations in regards to mortality rates as compared to conventional AMB.89–93 Harbarth et al86 performed a study which showed improve mortality rate with the use of Abelcet® as compared to conventional AMB which may have been attributable to reduced nephrotoxicity rates. In addition, one meta-analysis of all three nanoformulations as compared to conventional AMB demonstrated a significantly reduced risk of all-cause mortality (odds ratio [OR] 0.72; 95% confidence interval [CI] 0.54–0.97).94
Clinical cure
Several studies have shown similar clinical cure rates (confirmed microbiological cases) with nanoformulations of AMB as compared to conventional AMB.89–94 In the case of fungal infections, clinical cure becomes difficult to measure due to the ineffective microbiological growth. Due to low rates of confirmed fungal cases in reported studies, the clinical cure rate has not been found to be significantly different between the nanoformulation products and conventional AMB as statistical power was not likely met.
Nephrotoxicity
Based on numerous trials, the most dramatic and beneficial effect of all three nanoformulations appears to be the decreased rate of nephrotoxicity as compared to conventional AMB.86,89,91,92,94,95,97 The definition of nephrotoxicity varies amongst studies and may be reported as a doubling of serum creatinine, an increase of serum creatinine by at least 1 mg/dL, or a 50% decrease in calculated creatinine clearance. A meta-analysis done by Barrett et al94 demonstrated reduced nephrotoxicity (doubling of serum creatinine) by 58% with both Abelcet® and AmBisome® compared with conventional AMB (OR 0.42; 95% CI 0.33–0.54), while a study conducted by White et al97 demonstrated a 77% reduction in nephrotoxicity (composite score of change in serum creatinine) with Amphotec® as compared to conventional AMB (OR 0.23; 95% CI 0.12–0.43). It should be noted that the nanoformulations have not been directly compared to each other; thus, it is unknown if one agent is superior in the amount of renal protection provided in the same patient population.
Infusion rate
Two of the nanoformulations, Abelcet® and AmBisome®, allow a faster infusion rate (a 2-hour infusion for maximal dosing) than either Amphotec® or conventional AMB (a 5-hour infusion for maximal dosing). According to the product information provided in 1997 by Fujisawa USA Inc., the infusion time with AmBisome® may be reduced even further to a minimum of 1 hour in tolerant patients. The necessity to prolong infusion time is secondary to high rates of infusion-related reactions (chills, rigors, fever) associated with Amphotec® and conventional AMB. According to the product information provided in 1996 by The Liposome Company, Amphotec® has even higher rates of infusion related reactions than conventional AMB. The infusion-related reactions are generally most severe with the first dose and generally decline with time. The more convenient formulations (Abelcet® and AmBisome®) may allow for decreased home health-care time as well as freeing IV lines for administration of other necessary therapies while hospitalized.
Other ADRs
The nanoformulations appear to have similar rates of other adverse effects (not mentioned above) compared to conventional AMB (incidence of 5%–10% listed): nausea/vomiting, dyspnea, tachycardia, and hypokalemia (reports range up to 50% due to varying definitions).
Cost
The cost of the new nanoformulations is dramatically increased compared to the cost of conventional AMB (Table 2). Cost analyses are complex and difficult to truly assess when including non-pharmacy related expenditures such as AKI which may incur dialysis costs and those associated with prolonged length of stay; mortality may also appear to decrease costs due to shortened length of stay. A retrospective analysis98 reviewed 707 charts to determine the cost of renal failure associated with conventional AMB that resulted in an increased total cost of $29,823/patient who developed AKI. When this cost was averaged out amongst all study participants, the increased cost of conventional-AMB-associated renal failure was $8,947 (or $596 per day). Of note, like all retrospective reviews, causality of renal failure cannot be determined due to confounding contributory variables that may have existed. Based on this information, the nanoformulations may be more cost-effective than conventional AMB when the cost of the product is less than approximately $600/day. An even more practical approach may be to only use conventional AMB in patients who have low risk of developing AKI (ie, younger patients without other concomitant nephrotoxic agents and who have normal baseline renal function).
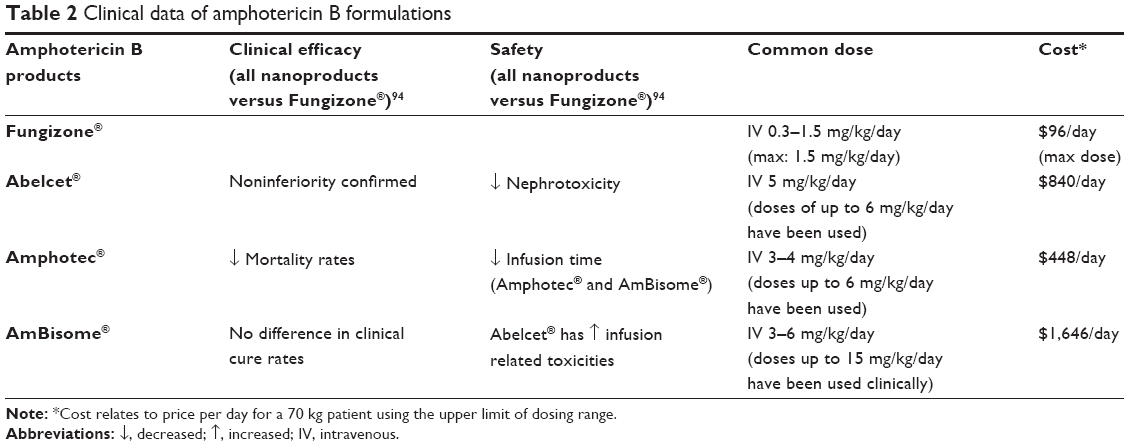 | Table 2 Clinical data of amphotericin B formulations |
Summary about AMB formulations
AMB has three nanoformulations which have been developed and studied as compared to conventional AMB. Of all commercially available nanoproducts, AMB has probably been one of the most robustly studied and reviewed as far as clinical application. It appears that all three nanoformulations have been proven to be as effective as conventional AMB and to potentially improve mortality rates, but not clinical cure rates, compared to the use of conventional AMB. Conventional AMB dosing is limited due to high rates of nephrotoxicity. The potentially improved mortality rates observed with the nanoformulations may be directly attributable to the ability to use higher doses for the long duration necessary to treat invasive systemic fungal infections by avoiding the potential nephrotoxicity caused by conventional AMB. Two of the nanoformulations (Abelcet® and AmBisome®) also allow faster infusion rates than conventional AMB with less infusion-related reactions, thus potentially decreasing home health-care time and freeing IV lines in the inpatient setting.
While it appears that the AMB nanoformulations have proven themselves an important development for the treatment of invasive fungal infections, they have not been directly compared to each other. Until such studies have been conducted, the determination of which agent to use should be based on the availability and cost to the institution. Based on our review, the use of the AMB nanoformulations, when and where available, is recommended over the use of conventional AMB in patients at highest risk for developing AKI (patients on concomitant nephrotoxic agents and/or increased baseline serum creatinine levels).
Fenofibrate formulations
Overview
Fenofibrate, marketed under various trade names, is a popular antilipidemic medication known for its poor oral bioavailability, requiring it to be dosed with food. Several dosage forms of fenofibrate are currently available (Table 3). Each aims to increase the bioavailability of the medication, primarily through smaller particle size, thus eliminating the need to be dosed with meals. The current formulation of Tricor® and Triglide® are nanoparticle formulations of fenofibrate. Fenofibrate is a peroxisome proliferator receptor alpha activator and is used as an adjunct to diet for the treatment of hypercholesterolemia and hypertriglyceridemia (product information for Tricor, Abbott Laboratories January 2014). Fenofibrate is an inactive prodrug that once hydrolyzed forms fenofibric acid.99 Fenofibric acid reduces total cholesterol (TC), low-density lipoprotein cholesterol (LDL), triglycerides (TG), and very-low-density lipoprotein concentrations; and increases high density lipoprotein cholesterol (HDL). This class of antilipidemics, commonly referred to as fibrates, also includes gemfibrozil (brand name Lopid®) and clofibrate (now discontinued). Fenofibrate is known for fewer ADRs and drug interactions than others in the fibrate class. Chemically, fenofibrate is a lipophilic and poorly soluble compound; therefore, maintaining therapeutic blood levels is difficult. In its original formulation, fenofibrate displayed poor bioavailability, which increased by 35% when taken with food.37 Meals containing higher fat levels may further increase absorption of fenofibrate by acting as an emulsifier to increase its solubility.37 Therefore, patients are counseled to take this fenofibrate formulation with meals, possibly negatively affecting compliance and subsequent lipid control. Various manufacturers of fenofibrate have aimed to improve formulations by decreasing particle size in efforts to increase solubility and bioavailability, therefore eliminating administration constraints.99
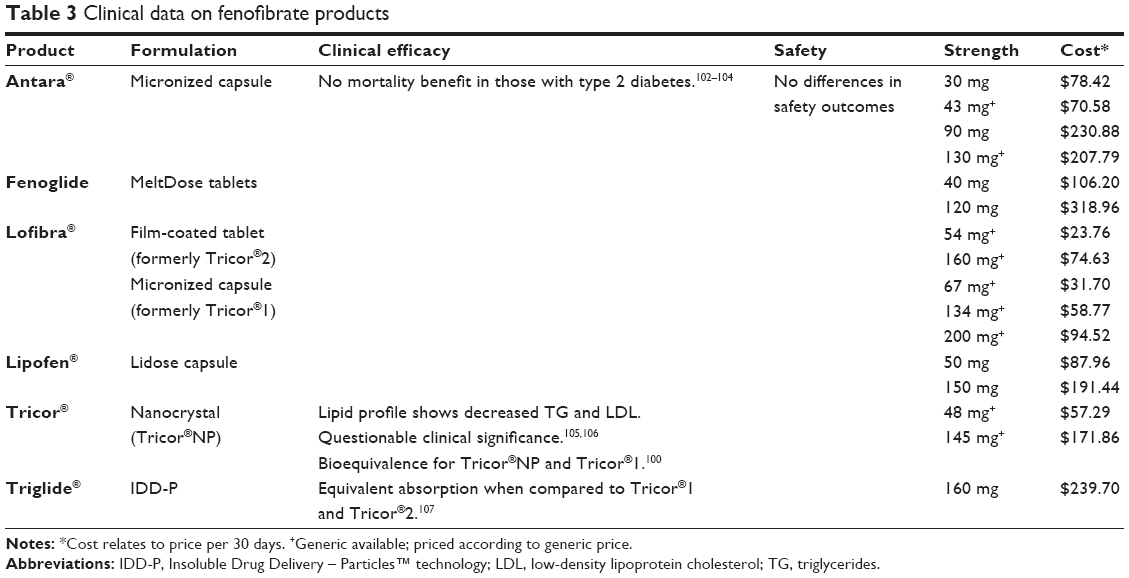 | Table 3 Clinical data on fenofibrate products |
Table 3 lists all currently available fenofibrate formulations. The brand-named Tricor® has been reformulated and rereleased twice since its original 1998 micronized capsule formulation (Tricor®1). In 2001, a micronized tablet (Tricor®2) replaced the capsule and in May 2004, Abbott Laboratories (Abbott Park, IL, USA) released Tricor® 145 mg fenofibrate using nanoparticle technology (Tricor®NP).100 This latest formulation does not have food restrictions, potentially increasing patient compliance and blood levels of fenofibrate.101
Abbott Laboratories, the maker of all formulations of Tricor®, is under legal scrutiny due to the alleged “subtle” reformulations of fenofibrate and the extension of the Tricor® brand.100 Through the continued release of branded formulations, Abbott Laboratories dominates the fenofibrate market and prevents the substitution to a generic product.100
Clinical efficacy: lipid lowering
Maciejewski and Hilleman completed a retrospective medical record review to compare the effectiveness of Tricor®NP 145 mg to Tricor®2 160 mg in patients with dyslipidemia and coronary heart disease.105 Subjects included in the review (n=130) must have been treated with Tricor®2 for at least 6 months prior to being switched to Tricor®NP. Subjects must have continued Tricor®NP for at least 3 months. Subjects were analyzed according to concomitant statin use. For each fenofibrate formulation, LDL, HDL, and TG levels were analyzed. Statistically significant differences were observed in LDL and TG levels, but not in HDL for both groups. In the group on combined fenofibrate and statin therapy, the authors reported a 2.8% reduction in LDL (P=0.002) and a 5.1% reduction in TG (P<0.0001) with the Tricor®NP compared to those on Tricor®2. In subjects on fenofibrate alone, the authors reported a decrease in LDL of 2.3% (P=0.009) and TG of 4.6% (P=0.0008) after the switch to Tricor®NP. The authors conclude that Tricor®NP is associated with greater improvements in LDL and TG compared to the original formulation, which may be attributed to increased bioavailability of the nanoparticle formulation.105
Davidson and Jones used electronic medical records to retrospectively compare TC, LDL, HDL, and TG levels in subjects with a history of hypertension, dyslipidemia, or diabetes.106 Subjects (n=491) must have taken Tricor®2 160 mg at least 60 days prior to switching to the Tricor®NP 145 mg. Statistically significant differences between Tricor®NP and Tricor®2 were found with TC (-5.5 mg/dL; P<0.0001), LDL (-5 mg/dL; P<0.0004), and TG (-6 mg/dL; P=0.004). No difference was found between groups with regard to HDL. Authors concluded that Tricor®NP offers a less restrictive dosing regimen that positively affects lipid outcomes.106
Finally, FDA approval studies of the Abbott Laboratories Tricor®NP showed bioequivalency between Tricor®NP 145 mg, three Tricor®NP 48 mg, and Tricor®1 200 mg under low-fat fed conditions in 68 healthy subjects.100,108 This was an open-label, single-dose, randomized, three-period, crossover study.108 While two of the studies show statistically significant decreases in LDL and TG with the nanoparticle formulation, it does not appear to be clinically significant. Also, lipid profiles serve as a surrogate marker only. The addition of the bioequivalency analysis suggests that, with equipotent dosing, similar lipid effects may be seen.
Fenofibrate formulated via IDD-P™ technology (Insoluble Drug Delivery – Particles™ technology) is called Triglide® and can be dosed without regard to food. Triglide® was compared to Tricor®1 200 mg and Tricor®2 160 mg in six pharmacokinetic studies in healthy volunteers. Triglide® showed equivalent absorption under fed and fasting conditions.107 Studies comparing the Triglide® with other fenofibrate formulations are lacking.
Mortality (cardiovascular studies)
Despite its actions on cholesterol, fenofibrate has not shown to reduce morbidity and mortality in type 2 diabetics. The FIELD study (Fenofibrate Intervention and Events Lowering in Diabetes) assessed the long-term coronary morbidity and mortality of Tricor®1 in type 2 diabetics. Participants (n=9,795) were randomly assigned to receive 200 mg Tricor®1 daily or placebo. Tricor®1 did not significantly reduce the risk of the primary outcome of coronary events as 5% in the treatment group and 6% in the placebo group experienced events (95% CI 0.75–1.05, P=0.16). Total mortality was also not reduced by Tricor®1 (7%) as compared to placebo (7%) (95% CI for difference 0.92–1.29, P=0.18). Of note, there was a high rate of statin use in the placebo group which may have altered the results.102
The NIH-funded ACCORD trial (Action to Control Cardiovascular Risk in Diabetes) evaluated the use of Tricor®2 in combination with simvastatin on morbidity and mortality in patients with type 2 diabetes. Subjects (n=5,518) were evaluated for an average of 4.7 years. The primary outcome, first occurrence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes, occurred in 2.2% in the fenofibrate group and 2.4% in the placebo group (hazard ratio 0.92; 95% CI 0.79–1.08, P=0.32). With respect to mortality, there was no statistically significant difference between groups (1.5% in fenofibrate group versus 1.6% in placebo; hazard ratio, 0.91; 95% CI 0.75–1.10, P=0.33).103
The results of the DAIS study (Diabetes Atherosclerosis Intervention Study) suggest that treatment with Tricor®1 200 mg daily reduces angiographic progression of coronary artery disease in type 2 diabetics. Although not statically powered to examine clinical endpoints, the fenofibrate group showed a pattern of reductions in cardiac endpoints.109 Due to the lack of evidence with respect to CV mortality, the role of fenofibrate in the treatment of dyslipidemia is severely limited. However, studies that have been conducted have not used the nanoparticle formulations of fenofibrate. The question remains: would a better-absorbed product change the outcome? Based off data currently available comparing formulations, it seems unlikely.
Fenofibrate is now primarily used in severe hypertriglyceridemia. While it is established that fenofibrate lowers triglyceride levels, its ability to prevent hypertriglyceride-induced pancreatitis has not been established.
Adverse reactions
There does not seem to be a difference in safety profiles between fenofibrate formulations. Common fenofibrate-associated ADRs are primarily gastrointestinal and include dyspepsia, abdominal pain, and nausea. Less common, but potentially more concerning ADRs include myopathy, abnormal liver function tests, and cholelithiasis. Fibrates, in general, may cause reversible increases in serum creatinine; however, this increase is not associated with renal damage.110,111
Cost
The nanoparticle formulation Triglide® is the most expensive of fenofibrate formulations and does not have a generic equivalent. Tricor® does have a less-expensive generic equivalent as do many of the other non-nanoparticle formulations.
Summary about fenofibrate formulations
There are few comparative studies showing safety and efficacy differences between the fenofibrate formulations. Also, FDA bioequivalence studies do not show any differences between Tricor® formulations, and there is a lack of data for Triglide®. As all products have similar side-effect profiles, the differences in formulation costs and food restrictions should be considered when selecting patient-specific treatment. While studies have shown a statistical improvement in intermediate lipid values, the clinical significance thereof is questionable.
Conclusion
Without any doubt, the launch of the NNI has and will have a significant impact on the development of new therapeutic, diagnostic, and/or theranostic approaches. The merger of nanoscience and nanotechnology with pharmaceutical research and development opens new horizons for the creation of novel drugs, which will utilize the unique characteristics of nanosized materials. The application of engineered nanomaterials to medicine will produce nanomedicines with unprecedented benefits for the clinical outcome of any potential therapeutic intervention.
In this article, we have examined over 40 clinically approved drugs which have been widely publicized as nanodrugs or nanopharmaceuticals. We argue that following the launch of the NNI in the year 2000, nanoscience terminology was quickly adopted by the pharmaceutical science community to an extent which we believe does not appear to be justified. We believe the true promise of nanoscience for drug development still has to materialize. This we will address in the second part of our review.
Acknowledgment
The authors gratefully acknowledge financial support provided by their institutions.
Disclosure
The authors report no conflicts of interest in this work.
References
Kreuter J. Colloidal drug delivery systems. 1st ed. Boca Raton, Ann Arbor, London, Tokyo: CRC Press; 1994. | ||
Babu A, Templeton AK, Munshi A, Ramesh R. Nanodrug delivery systems: a promising technology for detection, diagnosis, and treatment of cancer. AAPS Pharm Sci Tech. 2014;15(3):709–721. | ||
Bangham AD, Standish MM, Miller N. Cation permeability of phospholipid model membranes: effect of narcotics. Nature. 1965;208(5017):1295–1297. | ||
Bangham AD, Standish MM, Weissmann G. The action of steroids and streptolysin S on the permeability of phospholipid structures to cations. J Mol Biol. 1965;13(1):253–259. | ||
Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1):238–252. | ||
Marty JJ, Oppenheim RC, Speiser P. Nanoparticles – a new colloidal drug delivery system. Pharm Acta Helv. 1978;53(1):17–23. | ||
Forssen EA, Tökès ZA. Use of anionic liposomes for the reduction of chronic doxorubicin-induced cardiotoxicity. Proc Natl Acad Sci U S A. 1981;78(3):1873–1877. | ||
Forssen EA, Tökès ZA. Improved therapeutic benefits of doxorubicin by entrapment in anionic liposomes. Cancer Res. 1983;43(2):546–550. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine. 2013;9:1–14. | ||
Frequently asked questions [webpage on the Internet]. National Nanotechnology Initiative. Available from: http://www.nano.gov/nanotech-101/nanotechnology-facts. Accessed April 6, 2014. | ||
Liu X, Zhang Q, Tu Y, Zhao W, Gai H. Single gold nanoparticle localized surface plasmon resonance spectral imaging for quantifying binding constant of carbohydrate-protein interaction. Anal Chem. 2013;85(24):11851–11857. | ||
Schug TT, Johnson AF, Balshaw DM, et al. ONE Nano: NIEHS’s strategic initiative on the health and safety effects of engineered nanomaterials. Environ Health Perspect. 2013;121(4):410–414. | ||
Rivera Gil P, Hühn D, del Mercato LL, Sasse D, Parak WJ. Nanopharmacy: Inorganic nanoscale devices as vectors and active compounds. Pharmacol Res. 2010;62(2):115–125. | ||
Hiemenz JW, Walsh TJ. Lipid formulations of amphotericin B: recent progress and future directions. Clin infect Dis. 1996;22 Suppl 2:S133–S144. | ||
Juliano RL, Grant CW, Barber KR, Kalp MA. Mechanism of the selective toxicity of amphotericin B incorporated into liposomes. Mol Pharmacol. 1987;31(1):1–11. | ||
Juliano RL, Daoud S, Krause HJ, Grant CW. Membrane-to-membrane transfer of lipophilic drugs used against cancer or infectious disease. Ann N Y Acad Sci. 1987;507:89–103. | ||
Eckardt JR, Campbell E, Burris HA, et al. A phase II trial of DaunoXome, liposome-encapsulated daunorubicin, in patients with metastatic adenocarcinoma of the colon. Am J Clin Oncol. 1994;17(6):498–501. | ||
Guaglianone P, Chan K, DelaFlor-Weiss E, et al. Phase I and pharmacologic study of liposomal daunorubicin (DaunoXome). Invest New Drugs. 1994;12(2):103–110. | ||
Forssen EA, Malé-Brune R, Adler-Moore JP, et al. Fluorescence imaging studies for the disposition of daunorubicin liposomes (DaunoXome) within tumor tissue. Cancer Res. 1996;56(9):2066–2075. | ||
Kim S, Kim DJ, Geyer MA, Howell SB. Multivesicular liposomes containing 1-beta-D-arabinofuranosylcytosine for slow-release intrathecal therapy. Cancer Res. 1987;47(15):3935–3937. | ||
Glantz MJ, Jaeckle KA, Chamberlain MC, et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin Cancer Res. 1999;5(11):3394–3402. | ||
Alam M, Hartrick CT. Extended-release epidural morphine (DepoDur): an old drug with a new profile. Pain Pract. 2005;5(4):349–353. | ||
Pasero C, McCaffery M. Extended-release epidural morphine (DepoDur). J Perianesth Nurs. 2005;20(5):345–350. | ||
Northfelt DW, Martin FJ, Working P, et al. Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma. J Clin Pharmacol. 1996;36(1):55–63. | ||
Herzog C, Hartmann K, Kunzi V, et al. Eleven years of Inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine. 2009;27(33):4381–4387. | ||
Mischler R, Metcalfe IC. Inflexal V a trivalent virosome subunit influenza vaccine: production. Vaccine. 2002;20 Suppl 5:B17–B23. | ||
Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–796. | ||
Frampton JE. Mifamurtide: a review of its use in the treatment of osteosarcoma. Paediatr Drugs. 2010;12(3):141–153. | ||
Batist G, Barton J, Chaikin P, Swenson C, Welles L. Myocet (liposome-encapsulated doxorubicin citrate): a new approach in breast cancer therapy. Expert Opin Pharmacother. 2002;3(12):1739–1751. | ||
Ichikawa K, Takeuchi Y, Yonezawa S, et al. Antiangiogenic photodynamic therapy (PDT) using Visudyne causes effective suppression of tumor growth. Cancer Lett. 2004;205(1):39–48. | ||
Janknegt R, de Marie S, Bakker-Woudenberg IA, Crommelin DJ. Liposomal and lipid formulations of amphotericin B. Clinical pharmacokinetics. Clinical Pharmacokinet. 1992;23(4):279–291. | ||
de Marie S, Janknegt R, Bakker-Woudenberg IA. Clinical use of liposomal and lipid-complexed amphotericin B. J Antimicrob Chemother. 1994;33(5):907–916. | ||
Hershfield M. Adenosine Deaminase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993–2014:online. | ||
Davis FF. The origin of pegnology. Adv Drug Deliv Rev. 2002;54(4):457–458. | ||
Wang R, Billone PS, Mullett WM. Nanomedicine in Action: An overview of cancer Nanomedicine on the market and in Clinical Trials. J Nanomater. 2013;2013:1–12. | ||
Junghanns JU, Muller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3(3):295–309. | ||
Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25–31. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27(12):2569–2589. | ||
Chertow GM, Burke SK, Lazarus JM, et al. Poly[allylamine hydrochloride] (RenaGel): a noncalcemic phosphate binder for the treatment of hyperphosphatemia in chronic renal failure. Am J Kidney Dis. 1997;29(1):66–71. | ||
Ishii H, Furuse J, Nagase M, Maru Y, Yoshino M, Hayashi T. A phase I study of hepatic arterial infusion chemotherapy with zinostatin stimalamer alone for hepatocellular carcinoma. Jpn J Clin Oncol. 2003;33(11):570–573. | ||
Masuda E, Maeda H. Antitumor resistance induced by zinostatin stimalamer (ZSS), a polymer-conjugated neocarzinostatin (NCS) derivative. I. Meth A tumor eradication and tumor-neutralizing activity in mice pretreated with ZSS or NCS. Cancer Immunol Immunother. 1995;40(5):329–338. | ||
Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet. 2003;42(13):1089–1105. | ||
Abraxane approved for metastatic pancreatic cancer. Cancer Discov. 2013;3:OF3. | ||
Saif MW. U.S. Food and Drug Administration approves Paclitaxel protein-bound particles (Abraxane®) in combination with gemcitabine as first-line treatment of patients with metastatic pancreatic cancer. JOP. 2013;14(6):686–688. | ||
Foote M. Using nanotechnology to improve the characteristics of antineoplastic drugs: improved characteristics of nab-paclitaxel compared with solvent-based paclitaxel. Biotechnol Annu Rev. 2007;13:345–357. | ||
Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. | ||
Foss FM. Interleukin-2 fusion toxin: targeted therapy for cutaneous T cell lymphoma. Ann N Y Acad Sci. 2001;941:166–176. | ||
Peng Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum Gene Ther. 2005;16(9):1016–1027. | ||
Gordon EM, Hall FL. Critical stages in the development of the first targeted, injectable molecular-genetic medicine for cancer. In: Kang C, editor. Gene Therapy Applications. InTech; 2011:461–492. | ||
Gordon EM, Hall FL. Rexin-G, a targeted genetic medicine for cancer. Expert Opin Biol Ther. 2010;10(5):819–832. | ||
Gregoriadis G. The carrier potential of liposomes in biology and medicine (second of two parts). N Engl J Med. 1976;295(14):765–770. | ||
Gregoriadis G. The carrier potential of liposomes in biology and medicine (first of two parts). N Engl J Med. 1976;295(13):704–710. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Allen TM, Hansen C, Rutledge J. Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta. 1989;981(1):27–35. | ||
Allen TM, Chonn A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 1987;223(1):42–46. | ||
Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–237. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756. | ||
Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta. 1992;1113(2):171–199. | ||
Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174(1–2):83–93. | ||
Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta. 1991;1068(2):133–141. | ||
Bawa R, Melethil S, Simmons WJ, Harris D. Nanopharmaceuticals: Patenting issues and FDA regulatory challenges. The Sci Tech Lawyer. 2008;5(2):1–6. | ||
Jones RG, Kahovec J, Stepto R, et al. Compendium of Polymer Terminology and Nomenclature: IUPAC Recommendations 2008 Edition. Cambridge: Royal Society of Chemistry; 2009. | ||
Veerareddy PR, Vobalaboina V. Lipid-based formulations of amphotericin B. Drugs Today (Barc). 2004;40(2):133–145. | ||
Davis FF, Van Es T, Palczuk NC. Non-immunogenic polypeptides. United States Patent US 41793371979. | ||
Elsadek B, Kratz F. Impact of albumin on drug delivery – new applications on the horizon. J Control Release. 2012;157(1):4–28. | ||
Lamy-Freund MT, Ferreira VF, Schreier S. Polydispersity of aggregates formed by the polyene antibiotic amphotericin B and deoxycholate. A spin label study. Biochim Biophys Acta. 1989;981(2):207–212. | ||
Higginbottom S, Meadows J. Anaesthetic formulations of propofol. WO 2003017977 A12003. | ||
Chiechi LM. Estrasorb. IDrugs. 2004;7(9):860–864. | ||
Mercola KE, Cline MJ. Sounding boards. The potentials of inserting new genetic information. N Engl J Med. 1980;303(22):1297–1300. | ||
Mercola KE, Stang HD, Browne J, Salser W, Cline MJ. Insertion of a new gene of viral origin into bone marrow cells of mice. Science. 1980;208(4447):1033–1035. | ||
Cline MJ. Perspectives for gene therapy: inserting new genetic information into mammalian cells by physical techniques and viral vectors. Pharmacol Ther. 1985;29(1):69–92. | ||
Gruss P, Khoury G. Gene transfer into mammalian cells: use of viral vectors to investigate regulatory signals for the expression of eukaryotic genes. Curr Top Microbiol Immunol. 1982;96:159–170. | ||
Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans – immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323(9):570–578. | ||
Gene Therapy Clinical Trials Worldwide: Journal of Gene Medicine Online Library [webpage on the Internet]. Hoboken: John Wiley and Sons Ltd.; 2013. Available from: http://www.wiley.com/legacy/wileychi/genmed/clinical/. Accessed May 2014. | ||
Wilson JM. Gendicine: the first commercial gene therapy product. Hum Gene Ther. 2005;16(9):1014–1015. | ||
Krimsky S. China’s gene therapy drug: do Shenzen SiBiono Gen-Tech’s claims hold up? Genewatch. 2005;18(6):10–13. | ||
Hall FL, Liu L, Zhu NL, et al. Molecular engineering of matrix-targeted retroviral vectors incorporating a surveillance function inherent in von Willebrand factor. Hum Gene Ther. 2000;11(7):983–993. | ||
Mandarano G, Lodhia J, Eu P, Ferris NJ, Davidson R, Cowell SF. Development and use of iron oxide nanoparticles (Part 2): The application of iron oxide contrast agents in MRI. Biomed Imaging Interv J. 2010;6(2):e13. | ||
Clément O, Siauve N, Cuénod CA, Frija G. Liver imaging with ferumoxides (Feridex): fundamentals, controversies, and practical aspects. Top Magn Reson Imaging. 1998;9(3):167–182. | ||
Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85(5):315–319. | ||
Prince MR, Zhang HL, Chabra SG, Jacobs P, Wang Y. A pilot investigation of new superparamagnetic iron oxide (ferumoxytol) as a contrast agent for cardiovascular MRI. J Xray Sci Technol. 2003;11(4):231–240. | ||
Howard DH. Effect of mycostatin and fungizone on the growth of Histoplasma capsulatum in tissue culture. J Bacteriol. 1960;79:442–449. | ||
Perlman D, Giuffre NA, Brindle SA. Use of Fungizone in control of fungi and yeasts in tissue culture. Proc Soc Exp Biol Med. 1961;106:880–883. | ||
Wingard JR, Kubilis P, Lee L, et al. Clinical significance of nephrotoxicity in patients treated with amphotericin B for suspected or proven aspergillosis. Clin Infect Dis. 1999;29(6):1402–1407. | ||
Harbarth S, Burke JP, Lloyd JF, Evans RS, Pestotnik SL, Samore MH. Clinical and economic outcomes of conventional amphotericin B-associated nephrotoxicity. Clin Infect Dis. 2002;35(12):e120–e127. | ||
Mehta J. Do variations in molecular structure affect the clinical efficacy and safety of lipid-based amphotericin B preparations? Leuk Res. 1997;21(3):183–188. | ||
Groll A, Giri N, Gonzalez C. Penetration of lipid formulations of amphotericin B into cerebrospinal fluid and brain tissue. 37th International Conference on Antimicrobial Agents and Chemotherapy; 1997; Toronto, Ontario. p. A90. | ||
Prentice HG, Hann IM, Herbrecht R, et al. A randomized comparison of liposomal versus conventional amphotericin B for the treatment of pyrexia of unknown origin in neutropenic patients. Br J Haematol. 1997;98(3):711–718. | ||
Walsh TJ, Anaissie EJ, Denning DW, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46(3):327–360. | ||
Leenders AC, Reiss P, Portegies P, et al. Liposomal amphotericin B (AmBisome) compared with amphotericin B both followed by oral fluconazole in the treatment of AIDS-associated cryptococcal meningitis. AIDS. 1997;11(12):1463–1471. | ||
Leenders AC, Daenen S, Jansen RL, et al. Liposomal amphotericin B compared with amphotericin B deoxycholate in the treatment of documented and suspected neutropenia-associated invasive fungal infections. Br J Haematol. 1998;103(1):205–212. | ||
Bowden R, Chandrasekar P, White MH, et al. A double-blind, randomized, controlled trial of amphotericin B colloidal dispersion versus amphotericin B for treatment of invasive aspergillosis in immunocompromised patients. Clin Infect Dis. 2002;35(4):359–366. | ||
Barrett JP, Vardulaki KA, Conlon C, et al. A systematic review of the antifungal effectiveness and tolerability of amphotericin B formulations. Clin Ther. 2003;25(5):1295–1320. | ||
Walsh TJ, Finberg RW, Arndt C, et al. Liposomal amphotericin B for empirical therapy in patients with persistent fever and neutropenia. National Institute of Allergy and Infectious Diseases Mycoses Study Group. N Engl J Med. 1999;340(10):764–771. | ||
Anaisse EJ, White MH, Uzun M. Abelcet (Amphotericin B lipid Complex) vs amphotericin B for treatment of invasive candidiasis: A prospective, randomized multi-center trial. 35th Interscience Conference on Antimicrobial Agents and Chemotherapy; San Francisco, California. CA1995, p. LM21. | ||
White MH, Bowden RA, Sandler ES, et al. Randomized, double-blind clinical trial of amphotericin B colloidal dispersion vs amphotericin B in the empirical treatment of fever and neutropenia. Clin Infect Dis. 1998;27(2):296–302. | ||
Bates DW, Su L, Yu DT, et al. Mortality and costs of acute renal failure associated with amphotericin B therapy. Clin Infect Dis. 2001;32(5):686–693. | ||
Ling H, Luoma JT, Hilleman D. A review of currently available fenofibrate and fenofibric acid formulations. Cardiol Res. 2013;4(2):47–55. | ||
Downing NS, Ross JS, Jackevicius CA, Krumholz HM. Avoidance of generic competition by Abbott Laboratories’ fenofibrate franchise. Arch Intern Med. 2012;172(9):724–730. | ||
Sauron R, Wilkins M, Jessent V, Dubois A, Maillot C, Weil A. Absence of a food effect with a 145 mg nanoparticle fenofibrate tablet formulation. Int J Clin Pharmacol Ther. 2006;44(2):64–70. | ||
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9,795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849–1861. | ||
ACCORD Study Group, Ginsberg HN, Elam MB, et al. Effects of combination lipid therapy in Type 2 Diabetes Mellitus. N Engl J Med. 2010;362:1563–1574. | ||
No authors listed. Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet. 2001;357(9260):905–910. | ||
Maciejewski S, Hilleman D. Effectiveness of a fenofibrate 145-mg nanoparticle tablet formulation compared with the standard 160-mg tablet in patients with coronary heart disease and dyslipidemia. Pharmacotherapy. 2008;28(5):570–575. | ||
Davidson MH, Jones PH. Retrospective comparison of the effectiveness of a fenofibrate 145 mg formulation compared with the standard 160 mg tablet. Clin Drug Investig. 2008;28(10):615–623. | ||
Guivarc’h PH, Vachon MG, Fordyce D. A new fenofibrate formulation: results of six single-dose, clinical studies of bioavailability under fed and fasting conditions. Clin Ther. 2004;26(9):1456–1469. | ||
Drug Approval Package: Tricor (fenofibrate) Tablets, Application No: 021656 [webpage on the Internet]. Silver Spring: U S Food and Drug Administration. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021656s000_TricorTOC.cfm2004. Accessed May 11, 2014. | ||
No authors listed. Effect of finofibrate on progression of coronary-artery disease in type 2 diabtees: the Diabetes Atherosclerosis Intervention Study, a randomized study. Lancet. 2001;357:905–910. | ||
Davidson MH, Armani A, McKenny JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol. 2007;99(6A):3C–18C. | ||
Berglud L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine society clinical practice guideline. J Endocrinol Metab. 2012;97:2969–2989. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.