")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Multicentric osteolysis with nodulosis, arthritis, and cardiac defect syndrome: loss of MMP2 leads to increased apoptosis with alteration of apoptotic regulators and caspases and embryonic lethality
Authors Mosig R, Schulz R, Kassiri Z, Schaffler M, Martignetti J
Received 10 July 2014
Accepted for publication 14 August 2014
Published 13 November 2014 Volume 2014:4 Pages 207—217
DOI https://doi.org/10.2147/AGG.S69675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Marc Glucksman
Rebecca A Mosig,1 Richard Schulz,2,3 Zamaneh Kassiri,4 Mitchell B Schaffler,5 John A Martignetti1
1Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Department of Pharmacology; 3Department of Pediatrics, 4Department of Physiology, Cardiovascular Research Centre, Mazankowski Alberta Heart Institute, University of Alberta, Edmonton, AB, Canada; 5Department of Biomedical Engineering, City College of New York, New York, NY, USA
Abstract: Inactivating mutations of matrix metalloproteinase 2 (MMP2) cause multicentric osteolysis with nodulosis and arthritis, one of a group of inherited osteolytic and arthritic disorders. We have previously shown that mice lacking Mmp2 share similar syndromic features with the human disorder, and at the cellular level, Mmp2-/- mouse osteoblasts and osteoclasts have reduced numbers and proliferation rates at critical developmental time points. While previously hypothesized, the effect of MMP2 loss on apoptosis has not been examined in this system. We therefore sought to clarify its role in mediating the developmental defects in Mmp2-/- mice using immunohistochemistry, immunoblot analysis, and quantitative reverse transcription polymerase chain reaction analysis. We also explored the effects of MMP2 inhibition in the osteogenic sarcoma cell line SaOS2. Loss of MMP2 resulted in increased apoptosis and caspase activation both in vitro and in vivo. MMP2-deficient cells had increased Fas expression and reduced levels of the key survival signals p-FAK, p-ERK, cFLIP, and Bcl-2. Notably, and in marked contrast to their original characterization, there was a significant increase in the in utero demise of homozygous Mmp2-/- embryos. Specifically, litters from heterozygous crosses consistently yielded nearly 85% fewer than expected homozygous Mmp2-/- pups. Taken together, our findings highlight a new role for MMP2 in preventing apoptosis during development and growth.
Keywords: MMP2, apoptosis, Fas, embryonic lethality, osteoblast
Introduction
Multicentric osteolysis with nodulosis and arthritis (MONA, MIM #259600) is one of a group of inherited “vanishing bone” disorders characterized by destruction of affected bones and joints that results from inactivating mutations in the matrix metalloproteinase 2 (MMP2) gene.1 Mmp2−/− mice provide a unique model for study of the human disease, in that they share key clinically defining features of MONA, including progressive loss of bone mineral density, articular cartilage destruction, and abnormal long bone and craniofacial development.2 These phenotypic changes are associated with marked and developmentally restricted in vivo decreases in osteoblast and osteoclast numbers.2 In part, decreased cell number in MMP2-deficient mice has been determined to be associated with decreased cellular proliferation rates.2 To further our understanding of how MMP2 loss results in this cellular phenotype and to return to a question we originally proposed, given the diverse role of MMP(s), we investigated whether there might also be a molecular mechanism by which MMP2-deficient osteoblasts undergo increased rates of apoptosis in addition to decreased rates of proliferation. Our studies demonstrate that through alteration of Fas and adhesion signaling cascades, MMP2-deficient osteoblastic cell lines underexpress antiapoptotic factors, resulting in increased apoptosis. These findings add to the understanding of the importance of MMP2 in cellular and organismal biology.
Materials and methods
SaOS2 cell culture and small interfering ribonucleic acid silencing
siMMP2 oligonucleotide (oligo) pools targeting human MMP2 were purchased from Dharmacon, Inc. Transfections were performed in serum-free media using Lipofectamine 2000 (Invitrogen) and 100 pmol of oligo per well of a 12-well plate as previously described.3 Stable siMMP2 clones were generated as previously described.4
Quantitative reverse transcription polymerase chain reaction
Ribonucleic acid (RNA) was collected (Qiagen RNeasy Mini Kit) and treated with DNase (Qiagen). A total of 0.5 μg of RNA was reverse transcribed per reaction using first-strand complementary deoxyribonucleic acid (DNA) synthesis with random primers (Iscript, Bio-Rad Laboratories).
Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems) using the primers listed in Table 1. Cycle number values were normalized with both glyceraldehyde 3-phosphate dehydrogenase and β-actin values. Values were analyzed as fold change compared with siLuciferase (non-targeting control oligonucleotide [siLuc]) or empty vector control values. Data shown are the average of three separate experiments, each done in triplicate. Statistical significance was determined by comparing fold change using the unpaired, two-tailed Student’s t-test assuming equal variances.
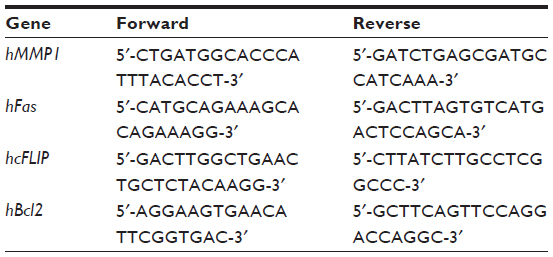 | Table 1 Primers used for quantitative reverse transcription polymerase chain reaction |
Adhesion assays
SaOS2 cells stably expressing siMMP2 oligos (siMMP2) or siLuc were plated at 150,000 cells per six-well matrix-coated plate in duplicate. All tissue culture plates and supplies were from BD Biosciences. Cells were photographed under a light microscope after 48 hours. Ten to 15 fields were photographed per well. Two independent experiments were performed to replicate results.
Fluoresence-activated cell sorting analysis
Cells were harvested from 12-well dishes and fixed in absolute ethanol (Sigma) for 24 hours. On the day of analysis, cells were pelleted and the absolute ethanol was removed. Cells were then stained with a 1 mL solution containing propidium iodide, RNase A, and phosphate-buffered saline (PBS). Fluoresence-activated cell sorting (FACS) analysis was performed on the FACSCalibur instrument (BD Biosciences).
Immunoblot analysis
SaOS2 cell extracts were harvested in radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology; standard protocol). Equal amounts of protein (50 μg; determined by the Bio-Rad DC protein quantification assay) were loaded and separated by PAGE and transferred to nitrocellulose membranes and probed with the specific antibodies listed in Table 2. Enhanced chemiluminescent immunoblot images were analyzed by scanning densitometry and quantified with Image J.5 Values were expressed as fold change relative to siLuc control.
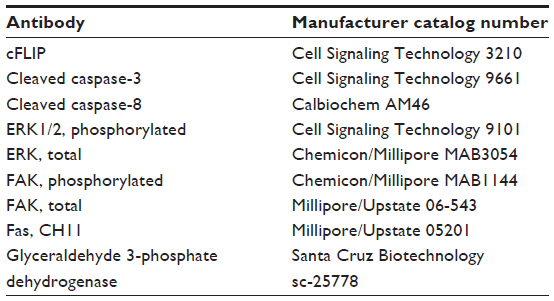 | Table 2 Antibodies used for immunoblots and immunohistochemical analysis |
Caspase-9 activity assay
Caspase-9 activation state was assayed using the ApoTarget Caspase-9 Colorimetric Protease Assay (Biosource International, Invitrogen #KHZ0101) according to the manufacturer’s protocol. Briefly, cells were harvested and lysed in the supplied lysis buffer and quantified for protein concentration. Samples were then diluted to 1 μg/μL. Reaction mixture and LEHD-pNA-modified substrate were incubated at 37°C for 2 hours protected from light. Results were read using a microplate reader at 400 nm. Background absorbance was subtracted and fold change over siLuc caspase-9 levels determined. Levels were assayed in triplicate in two separate experiments.
Animals
Generation of the original founder Mmp2−/− mice used in these studies was described by Itoh et al6 and was done by targeted deletion of the promoter, 5′UTR, and exon 1 of the Mmp2 gene. Genotyping was done by PCR of genomic DNA extracted from tail snips or yolk sacs as described previously.4
All procedures were performed in accordance with an institutionally approved protocol for the use of animals in research.
Histology and immunohistochemistry
Bones were isolated from age-matched Mmp2−/− and Mmp2+/+ mice and fixed in 10% phosphate-buffered formalin overnight and decalcified in cold ethylenediaminetetraacetic acid. Four micrometer-thick sagittal sections were stained with hematoxylin and eosin. Sections were stained with antibodies to cleaved caspase-3 (asp 175) and counterstained with hematoxylin. Two methods of negative controls were used. The first substituted PBS alone for primary antibody, and the second used a control rabbit serum in place of primary antibody. Positive cells were counted using Bioquant TCW software (Nashville, TN, USA) and normalized to the trabecular bone surface area. Forty tibia and 40 femur areas (measuring 700×550 μm each) were counted from 12 sections (six Mmp2−/− and six Mmp2+/+) (see Table 2 for antibodies used and manufacturers).
Embryo harvest
Pregnant females were sacrificed by cervical dislocation 7 or 9 days postcoitus. Embryos were isolated and photographed using a dissecting microscope under 4× objective. Yolk sacs were washed in sterile PBS and DNA isolated for genotyping analysis.
Results
Targeted small interfering ribonucleic acid-mediated MMP2 knockdown induces apoptosis in cultured cell lines
Our initial hypothesis that increased rates of cellular apoptosis may be a consequence of loss of MMP2 expression was drawn from the altered cell morphology we detected in SaOS2 osteoblast cells treated with small interfering RNA to MMP2. In adhesion assays of these siMMP2 osteoblasts, we observed an increased number of cells displaying a characteristic fragmentation and “blebbing” of the plasma membrane suggestive of apoptosis (Figure 1A). To directly test this hypothesis, we generated two stable cell lines, siMMP2-A and siMMP2-B. Both demonstrated >60% knockdown of the MMP2 messenger RNA (mRNA) transcript and MMP2 activity (Figure 1F). Interestingly, we did not identify clones with a greater degree of knockdown. In hindsight, the inability to generate cells with a greater degree of knockdown may be a consequence of the resultant cellular phenotype described herein. We assessed the amount of cell death in siMMP2 and control cells using propidium iodide staining for DNA content followed by FACS analysis to quantify cells in a subdiploid fragmented state (sub-G1). Remarkably, knockdown of MMP2 expression, even to these levels, resulted in three- to five-fold increased cell death compared with control cells (P<0.05, Figure 1B). We confirmed that increased cell death was induced through increased apoptosis, as opposed to necrosis or autophagy, by demonstrating a marked increase in caspase activation. Western blot analysis of cell lysates revealed over 30-fold increased levels of cleaved caspase-8 (p18) and caspase-3 (p17/19) in siMMP2 clones compared with siLuc control clones (Figure 1C and D, respectively). Caspase-9 activity was also markedly increased in both clones compared with controls (P<0.05, Figure 1E).
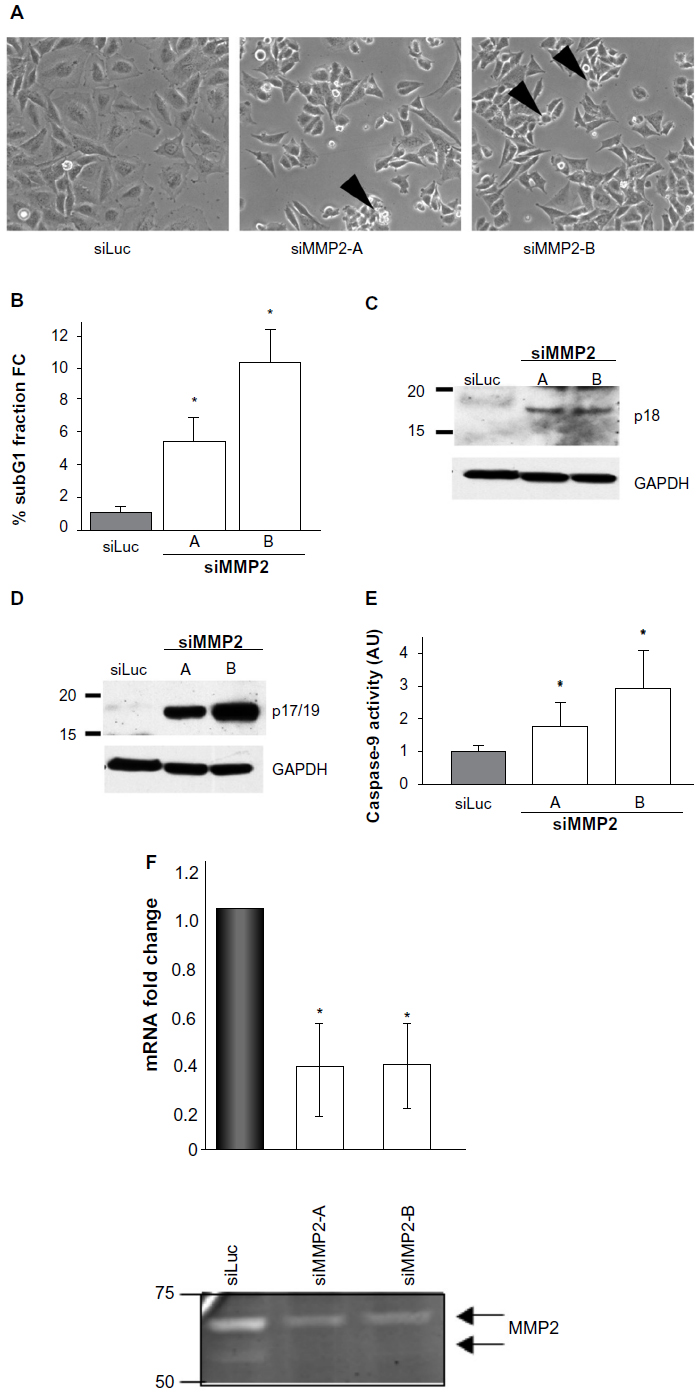 | Figure 1 MMP2-deficient SaOS2 clones demonstrate altered morphology, apoptosis, and caspase activation. |
Rates of cellular apoptosis are increased in perinatal Mmp2−/− mice
The marked degree of apoptosis induced in this ex vivo isolated cell culture system suggested a cell autonomous defect in the cells, possibly related to the cell autonomous defects we had previously identified in osteoblast differentiation and proliferation.2 This led us to investigate the in vivo level of apoptosis using Mmp2−/− mice. We had previously identified a reduced number of osteoblasts, osteoclasts, and proliferating cells in the bone marrow cavity of MMP2-deficient mice.2 This finding suggested a defect in proliferation (which we had previously demonstrated2), an increase in apoptosis, or a combination of both. To test the possibility that apoptosis was also affecting cell numbers, we examined the tibias and femurs of 4-day-old Mmp2−/− mice for cleaved caspase-3, a marker of active apoptosis.
There was an increase in cells positive for cleaved caspase-3 in the marrow cavities of the Mmp2−/− mice compared with those of control mice. Caspase-3-positive cells were mostly limited to the hypertrophic chondrocytes of the growth plate (arrows, Figure 2A and B). Microscopic examination revealed a greater than two-fold increase in the number of apoptotic lining cells along the endosteal and trabecular bone surface in the tibia, suggestive of an increased number of apoptotic osteoblasts (P<0.005, arrowheads, Figure 2A–C). By examining the number of positive staining osteocytes per bone area, a statistically significant upregulation of osteocyte apoptosis in both bones was demonstrated (Figure 2D, **P<0.005, *P<0.05, respectively). These findings of increased in vivo apoptosis suggested that the reduced number of osteoblasts 4 days after birth could be due to increased cell death.
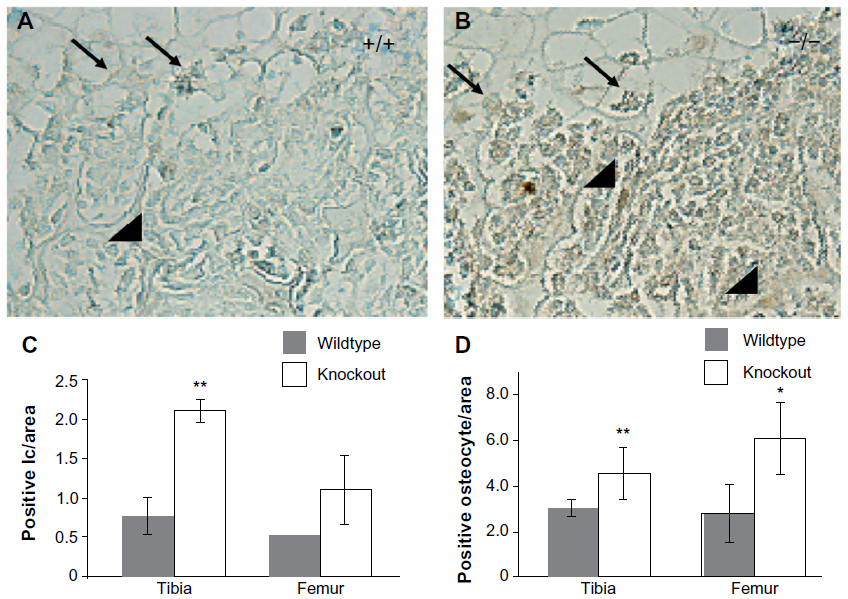 | Figure 2 Increased cleaved caspase-3 staining in matrix metalloproteinase 2 (Mmp2)−/− tibias. |
MMP2 inactivation alters activation of apoptotic and survival pathways
Having now identified increased levels of apoptosis both in vivo and in vitro in MMP2-deficient cells, we sought to investigate the potential mechanisms involved in the induction of apoptosis. We hypothesized that MMP2 could modulate death receptor pathway activation via cleavage of death receptors or their ligands. The Fas–FasL pathway was the primary candidate pathway because of its known strong effects on osteoblasts7 and because it has been shown to be affected by MMP2 loss in a number of other cell types.8–11 Furthermore, activation of both caspase-8 and caspase-9 in our siMMP2 SaOS2 clones demonstrated involvement of both the extrinsic and intrinsic apoptotic pathways, a hallmark of Fas-mediated apoptosis.11
We therefore examined the levels of Fas expressed by MMP2-deficient osteoblasts. Fas protein levels were found to be increased between two- and six-fold in siMMP2A and siMMP2B, respectively (Figure 3). Of particular interest, and in marked contrast to protein levels, Fas mRNA levels were not increased (Figure 3). This disjunction between RNA and protein levels suggested that the increased protein levels could be the result of posttranscriptional and/or posttranslational regulation. Moreover, Fas mRNA levels were decreased ~20% in the siMMP2-A clone compared with control (Figure 3, P<0.05). This suggested a selection for cells that have lower levels of Fas mRNA expression, as this would confer a survival advantage when there is altered Fas processing due to MMP2 loss. Fas generated in a bacterial cell expression system is sensitive to cleavage by MMP-7 but not MMP2.12 However, presentation of Fas on the cell surface, as well as variable levels of MMP-7 (barely detectable at the mRNA level in our control or siMMP2 SaOS2 cells) and MMP2, could affect Fas cleavage in osteoblastic cells.
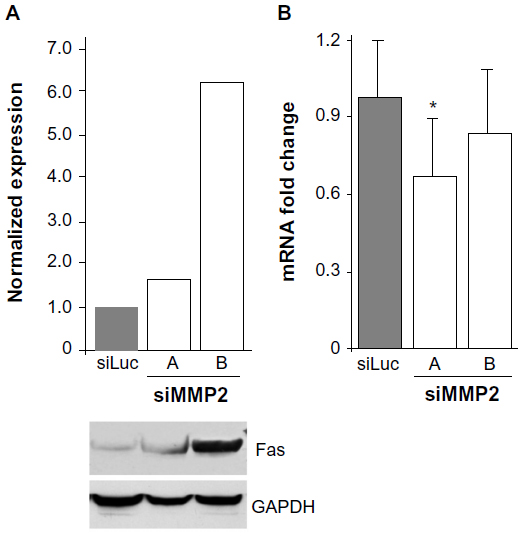 | Figure 3 Increased levels of Fas in MMP2-deficient cells. |
Activated caspase-3 is responsible for cleaving a large number of substrates, resulting in apoptotic propagation. Intriguingly, we had observed a decrease in cell adhesion in siMMP2 clones (data not shown). One particularly relevant candidate substrate potentially responsible for mediating this decreased adhesion phenotype is FAK. FAK is an important member of focal adhesions that form at attachment points between cells and the matrix.13 Following the induction of apoptosis, FAK is cleaved from its full-length 120 kDa active form to inactive 85 kDa and 77 kDa fragments.13 In siMMP2 clones, there are increased levels of these two cleavage products; the p85 fragment is increased ~10%–20% (Figure 4B), while the later degradation product, p77, is increased six- to eight-fold (Figure 4C).
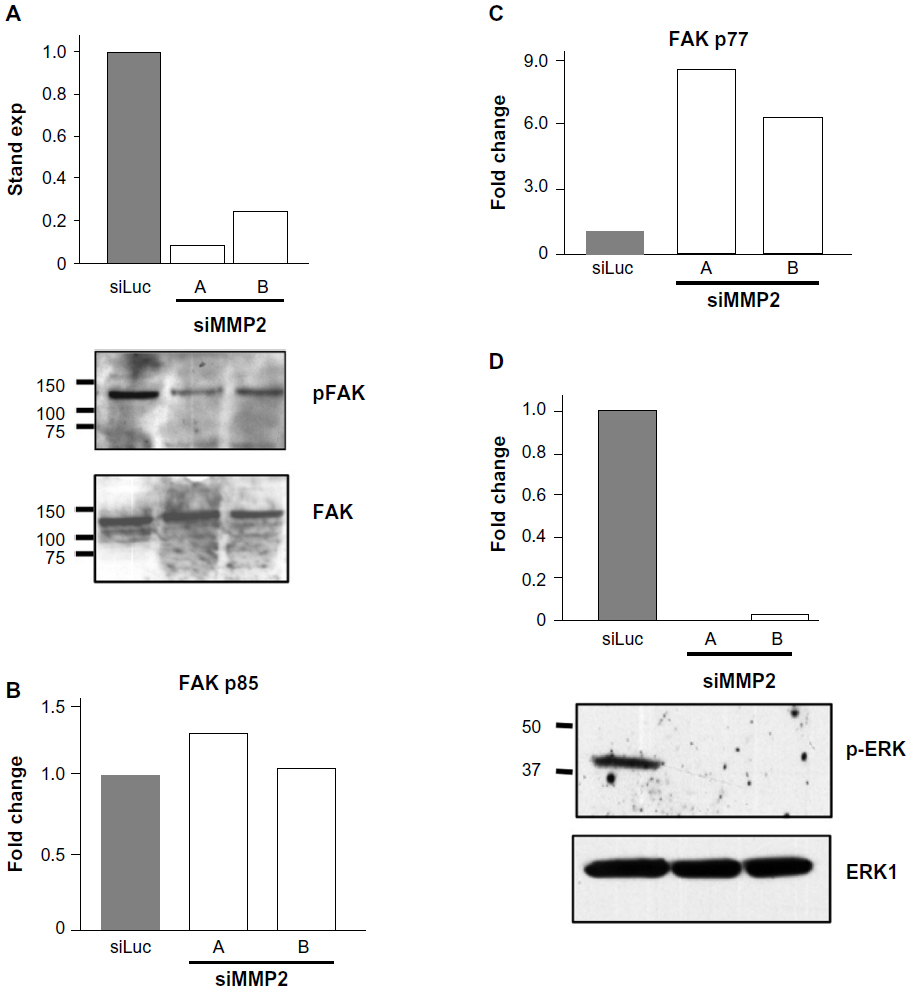 | Figure 4 Loss of FAK phosphorylation, increased FAK degradation, and loss of ERK phosphorylation in matrix metalloproteinase 2 (MMP2)-deficient cells. |
Phosphorylation of FAK takes place as part of the formation of focal adhesions as cells become attached to their matrix. We found that the phosphorylation status of FAK in siMMP2 clones is decreased by 70%–90% compared with the control cells (Figure 4A). Since ERK phosphorylation and activation are important for osteoblast survival and proliferation, we postulated that p-ERK would be affected in MMP2-deficient systems with aberrant FAK signaling, survival, and proliferation. In accord with these, ERK phosphorylation was reduced to almost nondetectable levels in siMMP2 clones (Figure 4D).
MMP2-deficient cells have decreased expression of the antiapoptotic proteins cFLIP and Bcl-2
Alterations in FAK and ERK signaling pathways may result in MMP2-deficient cells being more susceptible to Fas-mediated cell death via transcriptional changes. Transcription of the antiapoptotic caspase-8 inhibitor cellular FLICE-like inhibitory protein (cFLIP) is decreased by adhesion loss.14–16 cFLIP is an endogenous dominant negative inhibitor of caspase-8 (also known as Fas-linked interleukin 1β converting enzyme [FLICE]), and the balance between levels of caspase-8 and cFLIP can determine cellular susceptibility to death domain-mediated apoptosis.14 In siMMP2 SaOS2 clones, cFLIP levels were reduced at both the mRNA and protein levels (Figure 5). Specifically, levels of cFLIP transcript in siMMP2 clones were decreased approximately 50% (P<0.005, Figure 5A). The p48 isoform, cFLIP(S), which has the greatest inhibitory activity against caspase-8,17 was reduced by >70% at the protein level (Figure 5B).
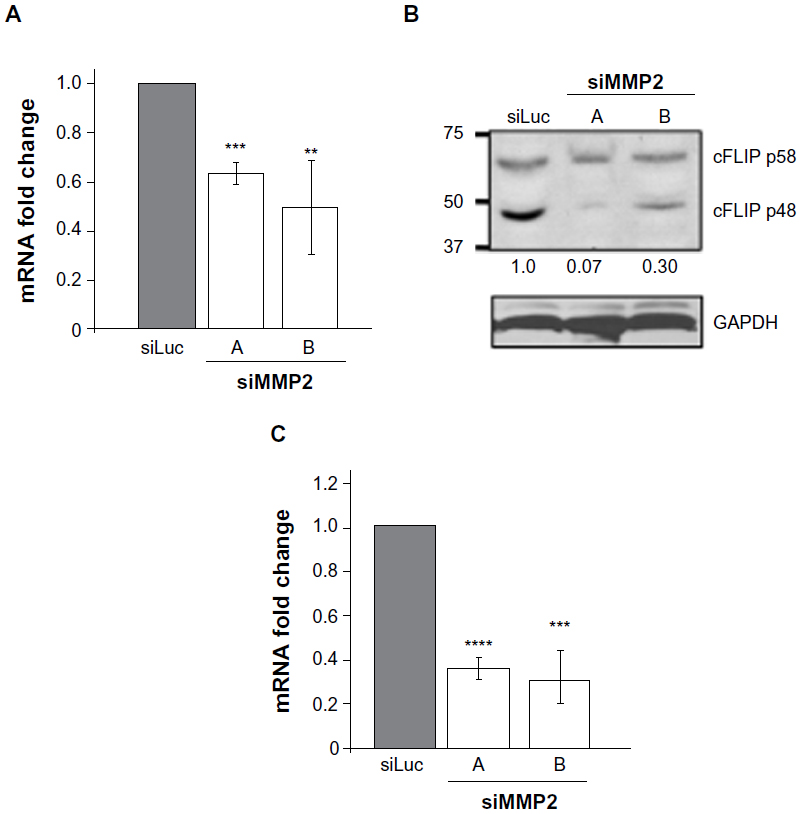 | Figure 5 Reduced levels of cFLIP and Bcl2 in MMP2-deficient cells. |
Bcl-2, which is responsible for inhibiting the depolarization of the mitochondrial membrane and cytochrome C release, can be upregulated in response to a number of differentiation and survival signals, particularly involving ERK activation.18 In agreement with the decreased level of ERK phosphorylation in MMP2-deficient cells, Bcl-2 mRNA expression levels were reduced >60% (P<0.0005, Figure 5C). The sum of these alterations in apoptotic regulators implies an increased susceptibility to Fas-mediated apoptosis.
MMP2-deficient cells have increased levels of Fas protein, as well as loss of antiapoptotic signaling pathways and proteins that may normally suppress apoptosis induced by Fas. These findings could explain the increased apoptosis in MMP2-deficient SaOS2 cells after activation of the Fas pathway, and in MMP2-deficient Jurkat cells (data not shown), which are known to be highly sensitive to Fas-mediated apoptosis.16,19
MMP2 deficiency results in reduced embryonic survival in mice
As we explored the mechanistic causes of increased apoptotic susceptibility, we began to synthesize a series of observations from the clinical and basic science aspects of MMP2 deficiency. First, our recent description of cardiac defects in multiple MONA families suggested that an extraskeletal phenotype may also be caused by MMP2 loss.20,21 In each of these families, a number of MMP2-deficient individuals were born with developmental heart defects, including septal and vessel defects. Second, the Fas, FAK, and ERK pathways and proteins affected by MMP2 loss are not specific to osteoblast function. They are key regulators of critical developmental pathways in multiple cell types and organ systems. Finally, and as fully detailed as follows, breeding of heterozygous mice results in fewer Mmp2−/− pups than expected. Taken together, these observations led us to reinvestigate the possibility of embryonic lethality in Mmp2−/− mice.
In our experiments, and in marked contrast to the original characterization of Mmp2−/− mice,6 litters from heterozygous crosses consistently yielded fewer than expected Mmp2−/− pups (Table 3). Based on expected numbers, there was an 85% reduction in the number of knockout pups born from heterozygote crosses. From 14 heterozygous crosses yielding a total of 104 live pups, only four Mmp2−/− pups were generated. This low number deviated markedly from the ~20 expected (χ2=22.5, P<0.0001). A number (n=23) of pups were found partially cannibalized; unfortunately, these could not be genotyped and their genetic status is unknown, limiting our functional analysis.
 | Table 3 Reduced number of matrix metalloproteinase 2 (MMP2)−/− pups from MMP2+/− heterozygous crosses |
To investigate the timing and plausible causes for this discrepancy, we examined the embryonic development of these mice. At both days E9.5 and E7.5, fewer knockout embryos were observed than expected. At E9.5, there was a 40% reduction in the expected number of knockout embryos from Mmp2+/− × Mmp2−/− crosses. Instead of the expected 1:1 ratio, there were only seven Mmp−/− embryos identified in the total of 23 embryos (χ2=3.522, P=0.06). In addition, surviving E9.5 Mmp2−/− embryos were dwarfed compared with Mmp2+/− embryos (Figure 6). As shown in Figure 6D, in some instances, Mmp2−/− embryos were chaotically disorganized and appeared as cell masses with no clearly recognizable embryonic features.
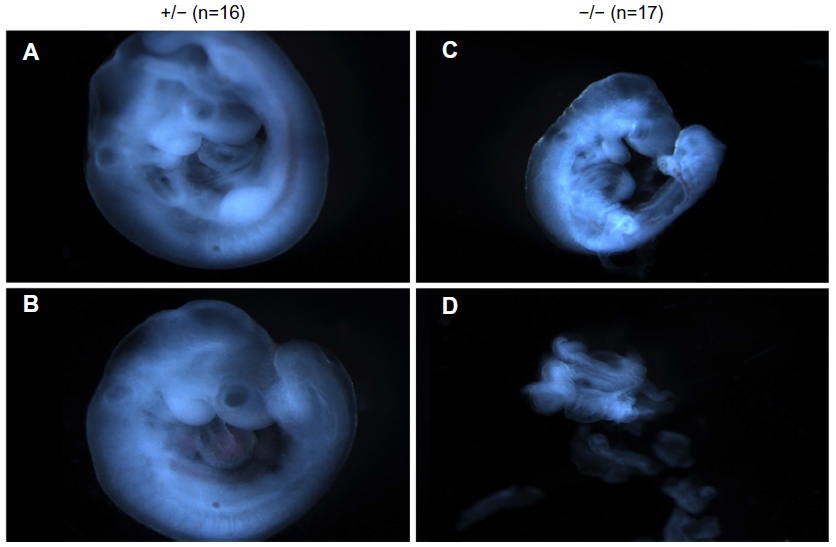 | Figure 6 Representative images of matrix metalloproteinase 2 (Mmp2)+/− (A, B) and Mmp2−/− (C, D) embryos at E9.5. |
At day E7.5, there was a 45% reduction in the expected number of Mmp2−/− embryos. Rather than the nine Mmp2−/− embryos expected from a total of 18 embryos, only five were Mmp2−/−(χ2=3.556, P=0.06). The surviving knockout embryos were shorter and had an average length of 689 μm compared with 827 μm for heterozygote embryos (Figure 7). This size difference, while not achieving statistical significance, was suggestive of a trend toward a growth defect (P=0.06, Figure 7C) and was in accord with the growth differences seen at day E9.5. Aside from smaller size, the Mmp2−/− embryos appeared morphologically normal.
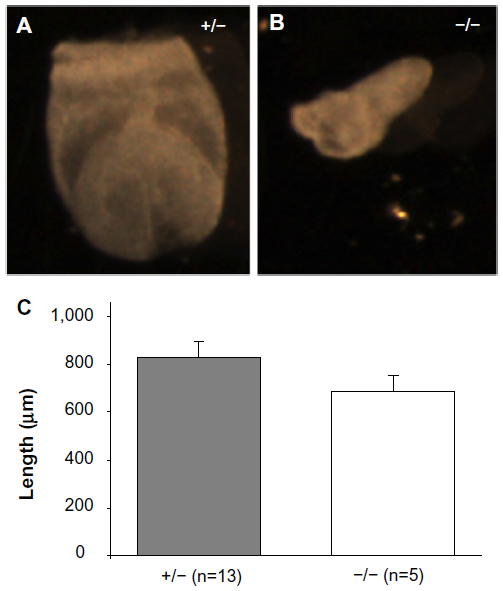 | Figure 7 Representative matrix metalloproteinase 2 (Mmp2)+/− (A) and Mmp2−/− (B) embryos at E7.5. (C) Lengthwise measurements of Mmp2−/− embryos revealed they were marginally shorter than Mmp2+/− embryos. |
Discussion
These results demonstrating a novel role for MMP2 in regulating apoptotic and survival pathways expand upon the current known function of MMP2 in modulating cell behavior. We have shown that loss of MMP2, both in vivo and ex vivo, induces cellular apoptosis. In vivo, the increased levels of apoptosis may contribute to the reduced viability of Mmp2−/− mouse embryos and pups as well as the skeletal features that define the surviving Mmp2−/− mice. We identified an increased level of Fas expression in siMMP2 clones. Furthermore, we demonstrated that loss of MMP2 causes decreased FAK and ERK phosphorylation and increased FAK degradation. These signaling aberrations could induce the decreased levels of two apoptosis inhibitors, cFLIP and Bcl-2, which we also identified in siMMP2 clones.
Our studies of MMP2 deficiency and the Mmp2−/− mouse identify a novel role for MMP2 as an antiapoptosis molecule and provide the mechanistic link and rationale for future studies. As our studies demonstrate, there is an increased number of apoptotic osteoblasts in the marrow cavity and bone of 4-day-old Mmp2−/− mice. Based on our molecular studies thus far, it appears this is the sum of removal of survival factors/signals (proper adhesion, FAK and ERK signaling, cFLIP and Bcl-2 expression) and inappropriate activation of apoptotic factors/signals (Fas signaling, caspase activation). It is difficult to isolate any one of these defects as being the most important or physiologically present in vivo. Of note, individual overexpression or knockout of a number of these factors results in abnormal skeletal phenotypes. Loss of integrin-1 disrupts cell adhesion and results in premature osteoarthritis.22 Caspase-3 deficiency, surprisingly, causes decreased bone formation and osteoblastogenesis.23 FasL-deficient mice that lack Fas signaling have increased bone mass and increased osteoblast colony formation, which represents the opposite phenotype to the Mmp2−/− mouse.24,25 Either Bcl-2 deficiency or overexpression affects osteoblast adhesion, differentiation, and apoptosis.26,27 Mice deficient in FAK, caspase-8, and cFLIP are all embryonic lethal. Therefore, their specific roles in skeletal development have not been studied.28–32 Future studies of Mmp2−/− mice should allow dissection of these changes and a greater understanding of their role in human disease and normal physiology.
Taken together, our data suggest a dual role for MMP2 in osteoblast biology. MMP2 facilitates adhesion as well as the removal of a “death” signal. Both of these actions could take place at the cell membrane. MMP2 activity could be targeted and concentrated pericellularly via the formation of complexes containing MMP2 and transmembrane proteins such as integrins or MT1-MMP.33–35 These interactions may originate intracellularly along the secretory pathway,36 which could explain why addition of exogenous MMP2 does not rescue the MMP2-deficient phenotype.2,4 Secondly, as MMP2 is released from the membrane into serum or conditioned media, it may remain in complex with TIMPs to prevent unregulated cleavage of matrix or serum proteins. This would further obstruct MMP2’s access and activity at the cell membrane upon addition to MMP2-deficient cells.
As cells adhere to a surface, MMP2, localized to the cell surface, may be responsible for cleavage, activation, or organization of either cell adhesion molecules or the matrix itself. In MMP2-deficient systems, these substrates go unprocessed, resulting in altered cell shape and adhesion. Within the cell, this may contribute to the reduced levels of phosphorylated FAK and ERK, which, in turn, could result in decreased levels of Bcl-2 and cFLIP. At the same time, MMP2 loss causes increased levels of Fas, resulting in increased Fas signaling.
Increased Fas activation is a primary candidate to explain the increased apoptosis in MMP2-deficient osteoblasts. Studies have shown that Fas activation in osteoblasts represses differentiation but does not induce apoptosis.7 However, this study was done in an MMP2-sufficient system. In MMP2-deficient cells, which are characterized by their reduced adhesion, reduced FAK and ERK activation, and reduced expression of Bcl-2 and cFLIP, the threshold for Fas-mediated apoptosis may be lower. Therefore, increased levels of Fas without these prosurvival signals would result in not just reduced differentiation but also increased apoptosis.
The role of MMP2 in cardiac development based on the finding of occasional cardiac defects in MONA patients20,21,37 and the known expression and role of MMP2 in rat and chicken cardiogenesis38,39 is, unfortunately, incomplete. Nonetheless, in these studies, it was noted that at the embryonic time points directly before and after looping of the embryonic heart tube to begin chambered heart development and septation there was a nearly equal reduction in the number of Mmp2−/− mice. It therefore appears that twisting of the heart tube is not directly affecting embryonic survival; rather, there is a more widespread disruption causing death prior to this point. This could be due to defects in implantation or other early stages, although we have not yet investigated this. Subsequent to cardiac looping, there is further loss of embryos, resulting in the highly significant loss of Mmp2−/− pups at birth. Therefore, it is possible that subsequent stages of cardiac development are affected by MMP2 loss. Studies to understand the causes of embryonic lethality, as well as the survival, of Mmp2−/− mice are ongoing.
The increased osteoblast cell death found in Mmp2−/− bones results in improper and incomplete bone modeling and remodeling early in life. Concomitantly, there is also decreased proliferation and adhesion of MMP2-deficient osteoblasts that may further disable matrix secretion and bone formation. The knowledge gained from these studies of MMP2 biology may contribute to a greater understanding of bone modeling and remodeling, osteoblast differentiation, and apoptosis in general.
Acknowledgments
We thank Drs Bruce Gelb and Cheryl Tan for their help and discussion with embryonic studies, the members of Dr Schulz’s and Kassiri’s laboratories for their efforts breeding mice and discussions of heart development, and Kelly Emerton in the Schaffler laboratory for her assistance with the ex vivo cleaved caspase-3 staining.
Disclosure
The authors report no conflicts of interest in this work.
References
Martignetti JA, Aqeel AA, Sewairi WA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28(3):261–265. | |
Mosig RA, Dowling O, Difeo A, et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16(9):1113–1123. | |
Narla G, DiFeo A, Yao S, et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65(13):5761–5768. | |
Mosig RA, Martignetti JA. Loss of MMP-2 in murine osteoblasts upregulates osteopontin and bone sialoprotein expression in a circuit regulating bone homeostasis Dis Model Mech. 2013;6(2):397–403. | |
Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. | |
Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 2007;272(36):22389–22392. | |
Kovacic N, Lukic IK, Grcevic D, Katavic V, Croucher P, Marusic A. The Fas/Fas ligand system inhibits differentiation of murine osteoblasts but has a limited role in osteoblast and osteoclast apoptosis. J Immunol. 2007;178(6):3379–3389. | |
Nyormoi O, Mills L, Bar-Eli M. An MMP-2/MMP-9 inhibitor, 5a, enhances apoptosis induced by ligands of the TNF receptor superfamily in cancer cells. Cell Death Differ. 2003;10(5):558–569. | |
Kargiotis O, Chetty C, Gondi CS, et al. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene. 2008;27(35):4830–4840. | |
Tsung AJ, Kargiotis O, Chetty C, et al. Downregulation of matrix metalloproteinase-2 (MMP-2) utilizing adenovirus-mediated transfer of small interfering RNA (siRNA) in a novel spinal metastatic melanoma model. Int J Oncol. 2008;32(3):557–564. | |
Chetty C, Bhoopathi P, Lakka SS, Rao JS. MMP-2 siRNA induced Fas/CD95-mediated extrinsic II apoptotic pathway in the A549 lung adenocarcinoma cell line. Oncogene. 2007;26(55):7675–7683. | |
Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stemenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61(2):577–581. | |
Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71(3–4):435–478. | |
Aoudjit F, Vuori K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: a role for c-Flip and implications for anoikis. J Cell Biol. 2001;152(3):633–643. | |
Shain KH, Landowski TH, Dalton WS. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol. 2002;168(5):2544–2553. | |
Yeh JH, Hsu SC, Han SH, Lai MZ. Mitogen-activated protein kinase kinase antagonized Fas-associated death domain protein-mediated apoptosis by induced FLICE-inhibitory protein expression. J Exp Med. 1998;188(10):1795–1802. | |
Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene. 2008;27(48):6216–6227. | |
Harnois C, Demers MJ, Bouchard V, et al. Human intestinal epithelial crypt cell survival and death: complex modulations of Bcl-2 homologs by Fak, PI3-K/Akt1, MEK/Erk, and p38 signaling pathways. J Cell Physiol. 2004;198(2):209–222. | |
Wen LP, Fahrni JA, Troie S, Guan JL, Orth K, Rosen GD. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272(41):26056–26061. | |
Tuysuz B, Mosig R, Altun G, Sancak S, Glucksman MJ, Martignetti JA. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 2009;17(5):565–572. | |
Castberg FC, Kjaergaard S, Mosig RA, et al. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur J Pediatr. 2013;172(12):1657–1663. | |
Zemmyo M, Meharra EJ, Kuhn K, Creighton-Achermann L, Lotz M. Accelerated, aging-dependent development of osteoarthritis in alpha1 integrin-deficient mice. Arthritis Rheum. 2003;48(10):2873–2880. | |
Miura M, Chen XD, Allen MR, et al. A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J Clin Invest. 2004;114(12):1704–1713. | |
Katavic V, Lukic IK, Kovacic N, Grcevic D, Lorenzo JA, Marusic A. Increased bone mass is a part of the generalized lymphoproliferative disorder phenotype in the mouse. J Immunol. 2003;170(3):1540–1547. | |
Kawakami A, Eguchi K, Matsuoka N, et al. Fas and Fas ligand interaction is necessary for human osteoblast apoptosis. J Bone Miner Res. 1997;12(10):1637–1646. | |
Boot-Handford RP, Michaelidis TM, Hillarby, et al. The Bcl-2 knockout mouse exhibits marked changes in osteoblast phenotype and collagen deposition in bone as well as a mild growth plate phenotype. Int J Exp Pathol. 1998;79(5):329–335. | |
Zhang W, Pantschenko AG, McCarthy MB, Gronowicz G. Bone-targeted overexpression of Bcl-2 increases osteoblast adhesion and differentiation and inhibits mineralization in vitro. Calcif Tissue Int. 2007;80(2):111–122. | |
Yeh WC, Pompa JL, McCurrach ME, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279(5358):1954–1958. | |
Yeh WC, Itie A, Elia AJ, et al. Requirement for casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12(6):633–642. | |
Varfolomeev EE, Schuchmann M, Luria V, et al. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9(2):267–276. | |
Ilic D, Furuta Y, Kanazawa S, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–544. | |
Ilic D, Kovacic B, Johkura K, et al. FAK promotes organization of fibronectin matrix and fibrillar adhesions. J Cell Sci. 2004;117(Pt 2):177–187. | |
Brooks PC, Stromblad S, Sanders LC, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85(5):683–693. | |
Galvez BG, Matias-Roman S, Yanez-Mo M, Sanchez-Madrid F, Arroyo AG. ECM regulates MT1-MMP localization with beta1 or alphavbeta3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J Cell Biol. 2002;159(3):509–521. | |
Deryugina EI, Ratnikov B, Monosov E, et al. MT1-MMP initiates activation of pro-MMP-2 and integrin alphavbeta3 promotes maturation of MMP-2 in breast carcinoma cells. Exp Cell Res. 2001;263(2):209–223. | |
Ali MA, Chow AK, Kandasamy AD, et al. Mechanisms of cytosolic targeting of matrix metalloproteinase-2. J Cell Physiol. 2012;227(10):3397–3404. | |
Gok F, Crettol LM, Alanay Y, et al. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur J Pediatr. 2010;169(3):363–367. | |
Linask KK, Han M, Cai DH, Brauer PR, Maisastry SM. Cardiac morphogenesis: matrix metalloproteinase coordination of cellular mechanisms underlying heart tube formation and directionality of looping. Dev Dyn. 2005;233(3):739–753. | |
Ratajska A, Cleutjens JP. Embryogenesis of the rat heart: the expression of collagenases. Basic Res Cardiol. 2002;97(3):189–197. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.