")
Back to Journals » OncoTargets and Therapy » Volume 8
Micheliolide overcomes KLF4-mediated cisplatin resistance in breast cancer cells by downregulating glutathione
Authors Jia Y, Zhang C, Zhou L, Xu H, Shi Y, Tong Z
Received 15 May 2015
Accepted for publication 30 July 2015
Published 28 August 2015 Volume 2015:8 Pages 2319—2327
DOI https://doi.org/10.2147/OTT.S88661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yongsheng Jia,1,* Chunze Zhang,2,* Liyan Zhou,1,* Huijun Xu,3 Yehui Shi,1 Zhongsheng Tong1
1Department of Breast Oncology, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Breast Cancer Prevention and Therapy, Ministry of Education, Key Laboratory of Cancer Prevention and Therapy, Tianjin Medical University, Tianjin, People’s Republic of China; 2Department of Colorectal Surgery, Tianjin Union Medicine Center, Tianjin, People’s Republic of China; 3Department of Oncology, Anhui Provincial Tumor Hospital, Hefei, People’s Republic of China
*These authors contributed equally to this work
Abstract: Micheliolide (MCL) is a promising novel compound with broad-spectrum anticancer activity. However, little is known regarding its action and mechanism in breast cancer. To explore the potential therapeutic application of MCL as a chemosensitivity modulator, this study investigated the effects of MCL on cisplatin sensitivity in breast cancer and the underlying mechanisms. In the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cytotoxicity assay and a xenograft tumor model, MCL enhanced the cisplatin sensitivity of the breast cancer cell line MCF-7 both in vitro and in vivo. Treatment of MCF-7 cells with low-dose cisplatin (10 µM) was sufficient to enrich the proportion of ALDH+ cells and upregulate Krüppel-like factor 4 (KLF4) expression. The results obtained from knockdown and overexpression experiments demonstrate that KLF4 is both necessary and sufficient to induce a cisplatin resistance phenotype in breast cancer cells. Furthermore, the glutathione (GSH) content was elevated in MCF-7 cells after overexpression of KLF4. KLF4-mediated resistance to cisplatin was found to be abrogated by treatment with buthionine sulfoximine, an inhibitor of GSH synthesis. MCL induced GSH depletion and severe cell death in KLF4-overexpressing MCF-7 cells following exposure to cisplatin. Therefore, these results suggest that MCL-mediated direct depletion of GSH represents a major mechanism in reversing KLF4-induced cisplatin resistance in MCF-7 cells.
Keywords: KLF4, cisplatin resistance, glutathione, breast cancer, Micheliolide
Introduction
The development of chemoresistance is a persistent problem during the treatment of advanced breast cancer, but the exact reasons underlying this process remain unclear. A growing number of reports have shown that a higher proportion of cancer stem cells (CSCs) correlate with an increased incidence of relapse due to chemotherapy-resistant outgrowth and worse overall survival. In addition, chemoresistant CSCs have the ability to reinitiate disease either immediately after treatment or after significant periods of dormancy. Therefore, targeting CSCs and CSC-related genes might provide a novel strategy to improve chemotherapeutic effects and suppress breast cancer recurrence.
Krüppel-like factor 4 (KLF4) is a zinc finger-type transcription factor that is required for both embryonic stem cell self-renewal and maintenance of pluripotency.1 In 2004, Pandya et al2 reported that the localization of KLF4 in the nucleus of breast cancer cells is a prognostic factor and identified KLF4 as a marker of an aggressive phenotype in early-stage infiltrating ductal carcinoma. Further evidence has demonstrated that KLF4 has a potent oncogenic role in mammary tumorigenesis likely by maintaining its stem cell-like features and by promoting cell migration and invasion.3 In 2014, Dong et al4 identified that KLF4 overexpression is associated with lower pathologic complete remission in patients with locally advanced breast cancer undergoing neoadjuvant chemotherapy. These studies suggest that KLF4 might provide an effective therapeutic approach to suppress tumorigenicity in breast cancer and might also serve as a predictor for pathologic complete remission in patients with breast cancer after neoadjuvant chemotherapy. In our previous study, we found that KLF4 regulates the cellular sensitivity to cisplatin in hepatocarcinoma stem-like cells and in hepatocarcinoma cells by elevating intracellular glutathione (GSH) levels.5 It was also demonstrated that there is an aberrant steady-state pool of GSH and a global upregulation of GSH pathway proteins in CSCs, which render them resistant to chemotherapy.6,7 On the basis of these findings, we hypothesized that KLF4 might also regulate the chemoresistance in breast cancer cells, and that the inhibition of GSH metabolism might provide a new therapeutic strategy for abrogating KLF4-mediated chemoresistance.
Parthenolide, a natural sesquiterpene lactone with a 10,5-ring structure isolated from Tanacetum parthenium, is a newly identified small molecule found to be selectively lethal against CSCs by targeting GSH metabolism pathways.2 Micheliolide (MCL), an orally bioavailable parthenolide analog that can also selectively eliminate CSCs, has only been reported to have been used in acute myelogenous leukemia6 and colorectal cancer.7 However, the detailed molecular mechanisms of the anticancer effects of MCL remain unresolved. To address these questions, the present study was designed to identify the potential effect of MCL in reversing KLF4-mediated cisplatin resistance and the related mechanisms in breast cancer cells.
Materials and methods
Cell culture and reagents
The human breast cancer cell line MCF-7, MDA-MB-231, and HEK293T cells were preserved by our laboratory, and MCF-7/VC3AI cells were kindly provided by Dr Binghui Li (Tianjin Medical University Cancer Institute and Hospital, Tianjin, People’s Republic of China). All cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone, Logan, UT, USA) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin mixture at 37°C in a humidified atmosphere of 5% CO2 and 95% air. All human cell line protocols were approved by the Ethics Committee of Tianjin Medical University.
Mammospheres culture
MCF-7 cells after treatment with cisplatin were suspended in serum-free DMEM (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with B27 (1:50, Thermo Fisher Scientific), 20 ng/mL hEGF (Thermo Fisher Scientific), 20 ng/mL basic fibroblast growth factor (R&D Systems, Inc., Minneapolis, MN, USA), 40 U/mL heparin (Sigma-Aldrich Co., St Louis, MO, USA), 2 mM glutamine (Sigma-Aldrich Co.), 100 U/mL penicillin, 100 μg/mL streptomycin, 5 μg/mL insulin (Sigma-Aldrich Co.), and 0.5 μg/mL hydrocortisone (Sigma-Aldrich Co.), and plated in 24-well ultra-low attachment plates (Corning Incorporated, Corning, NY, USA) at 2,000 cells per well. After 15–20 days, the plates were analyzed for mammospheres formation, and quantified using an inverted microscope (IX71; Olympus Corporation, Tokyo, Japan).
In vivo tumor growth experiments
Five- to 6-week-old female athymic BALB/c nude mice were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences. All animal protocols were approved by the Tianjin Medical University animal care committee. Subcutaneous tumor xenografts of human cancer cells were established in nude mice by injection of 2×106 cells in 100 mL 2 mg/mL Matrigel (VWR, Mississauga, ON, Canada) at both flanks near the posterior legs. Starting on the day of the xenografts, mice inoculated with hormone-dependent MCF-7 tumor cells received a subcutaneous injection of estradiol (E2) (0.15 mg/animal) at 3-day intervals, to prime tumor growth. For each animal, tumor size was measured twice a week using calipers. Tumor volume was calculated using the formula: (length × width2)/2. Treatments with the drugs were initiated when the tumors derived from a given cell line reached a mean volume of 150 mm3 (day 0). Mice received intraperitoneal injections of MCL (50 mg/kg), cisplatin (0.00615 mmol/kg), or control (vehicle) solution (0.9% sodium chloride in water), at 3-day intervals starting at day 0. Animals were also weighed twice a week starting at day 0. Mice were killed after 20 days; at that time, tumor size had not grown over 2,500 mm3.
Lentivirus production and transduction
Lentivirus production and titering were carried out according to the following protocols from Trono Lab (http://tronolab.epfl.ch): in HEK293T cells, the pLVTHM vector containing KLF4-shRNA (5′-GGA CGG CTG TGG ATG GAA A-3′)8 or the shRNA-negative control was co-transfected with the envelope plasmid pMD2.G and the packaging vector psPAX2 via calcium phosphate precipitation. Virus-containing medium was collected 48 hours after transfection, and the viruses were concentrated by ultra-centrifugation at 28,000 rpm for 2 hours. Concentrated viruses were reconstituted in phosphate-buffered saline (PBS). The lentiviruses pWPTS-KLF4 and pWPTS-GFP were produced as indicated previously.5
Cytotoxicity assay
Breast cancer cells were plated at a density of 0.8×104 cells per well in 96-well plates. After 24 hours incubation, the cells were treated with MCL or cisplatin at the indicated concentrations for 24 hours, then the medium was removed, and 200 μL fresh medium plus 20 μL MTT (5 mg/mL dissolved in PBS) was added to each well. After incubation for 4 hours at 37°C, the culture medium containing MTT was withdrawn, and 150 μL dimethyl sulphoxide was added followed by shaking for 10 minutes until the crystals were dissolved. Viable cells were detected by measuring the absorbance at 570 nm using an MRX II absorbance reader (DYNEX Technologies, Chantilly, VA, USA). The cell growth was expressed as the ratio of the absorbance of cells exposed to the MCL treatment to that of cells not exposed to the treatment (100%). The inhibition rate (IR) was calculated as follows:
|
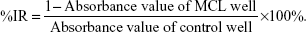
Western blot analysis
For western blot analysis, the cells were washed three times with PBS, homogenized in cell lysis buffer, incubated on ice for 20 minutes, and then centrifuged for 15 minutes at 10,000 rpm. The aqueous supernatant was collected and quantified using a BCA protein assay kit (Boster, Wuhan, People’s Republic of China). Equal amounts (30 μg) of protein extract were loaded and separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis. After electrophoresis, the proteins on the gel were transferred to a polyvinylidene fluoride membrane and incubated with the corresponding antibodies. The immune complexes were detected using the LI-COR Odyssey infrared imaging system (Li-Cor, Lincoln, NE, USA).
Analysis of apoptosis by flow cytometry
Cells (3×105) suspended in 2 mL fresh media were plated into each well of a 6-well, flat-bottomed microtiter plate and incubated overnight. Then, the cells were treated with or without 10 μM MCL for 24 hours. After being harvested, the cells were washed twice with pre-chilled PBS and then resuspended in 1× binding buffer at a concentration of 1×106 cells/mL. The solution (100 μL, 1×105 cells) was then mixed with 5 μL Annexin V-PE and 5 μL 7-AAD (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instruction. The mixed solution was incubated at room temperature away from light for 15 minutes, and then 400 μL 1× dilution buffer was added to each tube. Analysis was performed using a fluorescence-activated cell sorting flow cytometer within 1 hour.
Determination of intracellular GSH
Cells grown to 80% confluence in T-75 flasks were harvested with trypsin, washed in cold PBS, and resuspended in 0.154 M NaCl. Protein extracts were prepared by multiple freeze/thaw cycles and by sonication. These extracts were normalized for total protein content using a modified Bradford assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Total GSH was quantified with an enzymatic assay.9
Statistical analysis
All experiments were performed at least three times. Excel (Microsoft Corporation, Redmond, WA, USA) was used for statistical analysis, and statistical significance was determined using Student’s t-test. P<0.05 was considered as significant.
Results
MCL enhances the cisplatin sensitivity of MCF-7 cells in vitro and in vivo
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure the cytotoxic effects of cisplatin and MCL. Cells from the breast cancer cell line MCF-7 were plated and treated with MCL alone (5 or 10 μM), cisplatin alone (0, 2.5, 5, 10, 20, or 40 μM) or cisplatin at various concentrations combined with 5 or 10 μM MCL. After treatment for 24 hours (Figure 1A), treatment with MCL alone reduced cell viability and markedly enhanced the cytotoxic effects of cisplatin on MCF-7 cells.
 | Figure 1 MCL enhances the cisplatin sensitivity of MCF-7 cells in vitro and in vivo. |
Caspases are crucial mediators of apoptosis. Among them, caspase-3 is a frequently activated death protease, which catalyzes the specific cleavage of many key cellular proteins.10 Accordingly, cisplatin- and MCL-induced apoptosis were measured by monitoring real-time caspase-3-like activity in MCF-7/VC3AI cells obtained from the laboratory of Dr Binghui Li as previously described.11 As shown in Figure 1B, MCL significantly increased numbers of fluorescence-positive cells in MCF-7/VC3AI cells after treatment with cisplatin for 24 hours. Furthermore, the antitumor activity of the combination of MCL and cisplatin in nude mice xenografted with MCF-7 cells was higher than that induced by either agent alone (Figure 1C). This, together with the in vitro results, supports the notion that MCL causes greater sensitivity to cisplatin in the MCF-7 cells, which are relatively resistant to cisplatin treatment compared with other breast cancer cell lines.12
Cisplatin treatment elevates KLF4 expression levels
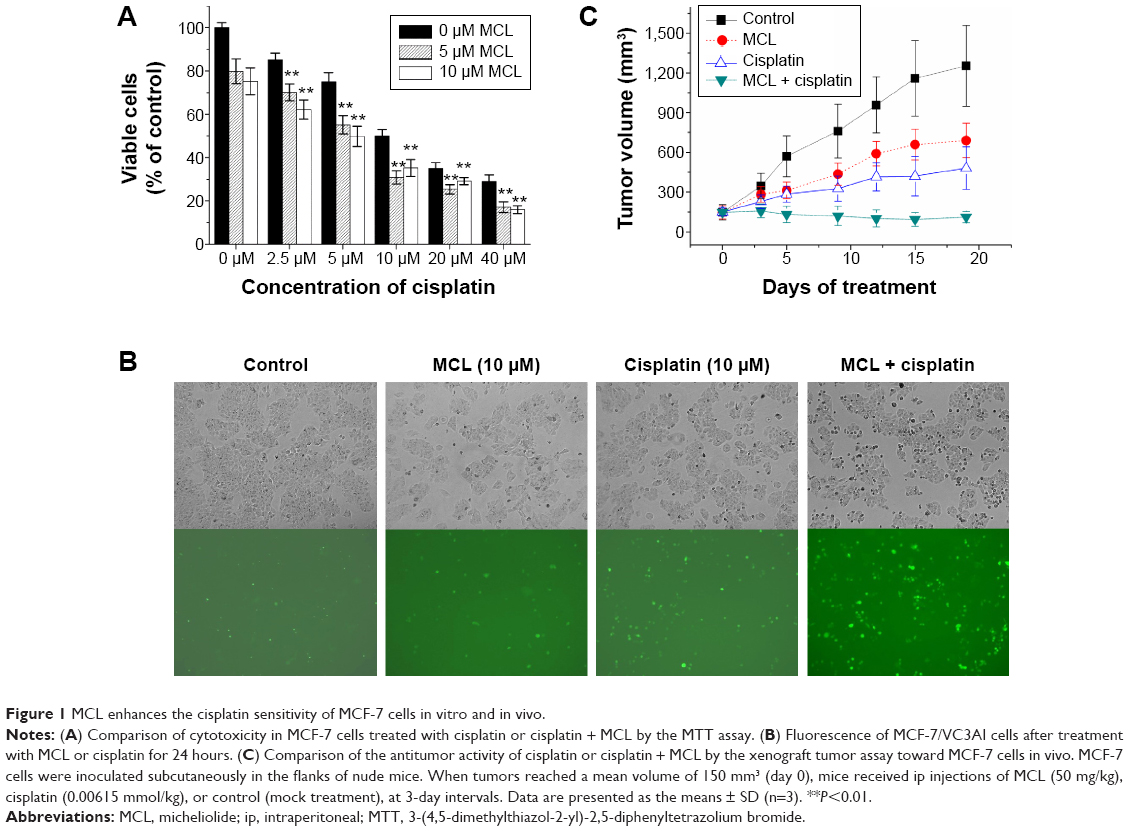
To clarify whether cisplatin might raise the percentage of CSCs in breast cancer tissue, we tested the cytotoxic effect of cisplatin in the MCF-7 cell line. As shown in Figure 2A, 10 μM cisplatin treatment for 24 hours led to a partial inhibition of MCF-7 cell survival. Following treatment, the remaining cells were subjected to a mammosphere assay. Compared with the controls, the cisplatin-treated cells generated more mammospheres (Figure 2B) and exhibited upregulation of the KLF4 protein (Figure 2C). Aldehyde dehydrogenase (ALDH) has been recognized as a marker of CSCs in breast cancer,13 and we found that cisplatin treatment also increased the ratio of ALDH+ cells in MCF-7 cell cultures (Figure 2D). As recent studies have suggested that CSCs might mediate chemoresistance, we surmised that the elevated expression of KLF4 might delineate a breast cancer phenotype that is highly resistant to cisplatin.
 | Figure 2 Cisplatin treatment elevates the ratio of ALDH+ cells and KLF4 expression in MCF-7 cells. |
Decreased sensitivity of breast cancer cells to cisplatin by KLF4
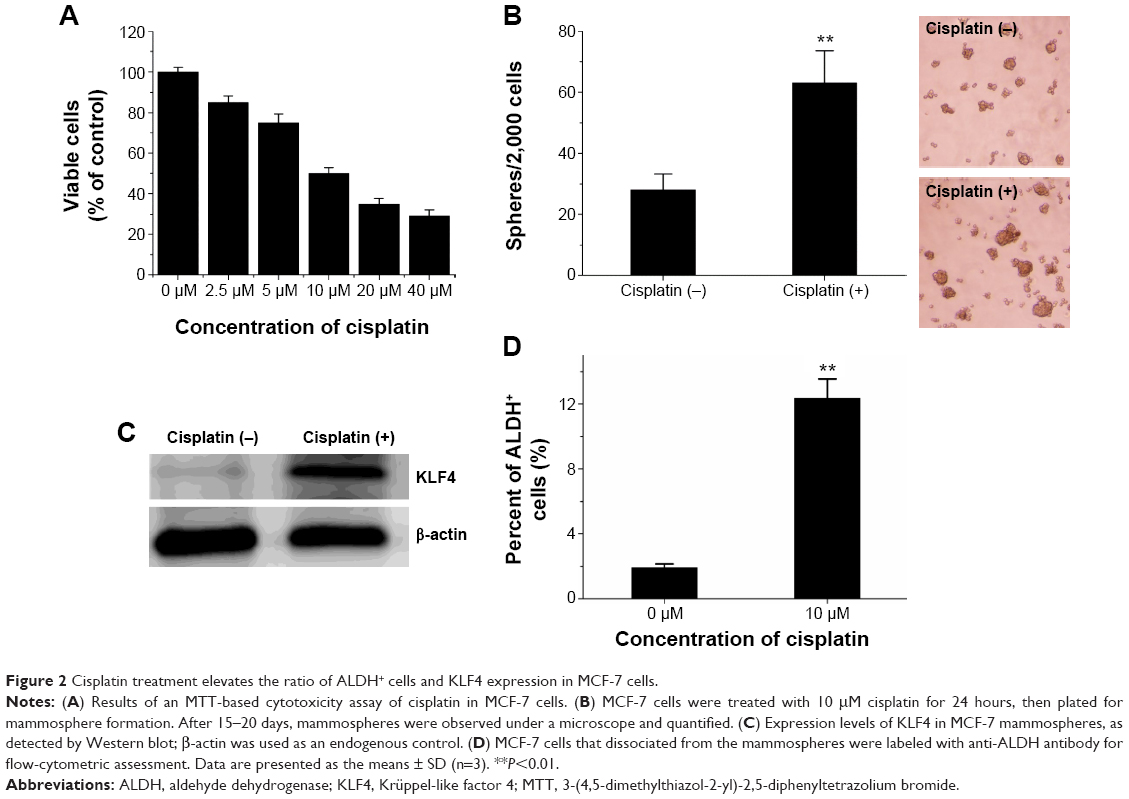
To determine the relationship between the elevated expression of KLF4 and cisplatin resistance in MCF-7 cells, MCF-7 cells were infected with pWPTS-KLF4 lentivirus to upregulate KLF4. As shown in Figure 3A, KLF4 was overexpressed in the pWPTS-KLF4-infected MCF-7 cells as determined by Western blot analysis. The results showed that MCF-7 cells infected with pWPTS-KLF4 had significantly enhanced resistance to cisplatin versus the controls (P<0.01) (Figure 3C). Accordingly, when KLF4 was knocked down by infection of lentivirus harboring KLF4-short hairpin RNA (shRNA) (Figure 3B), MDA-MB-231 cells became sensitized to cisplatin (P<0.01) (Figure 3D). Therefore, the sensitivity of breast cancer cells to cisplatin was determined to be negatively regulated by KLF4 levels.
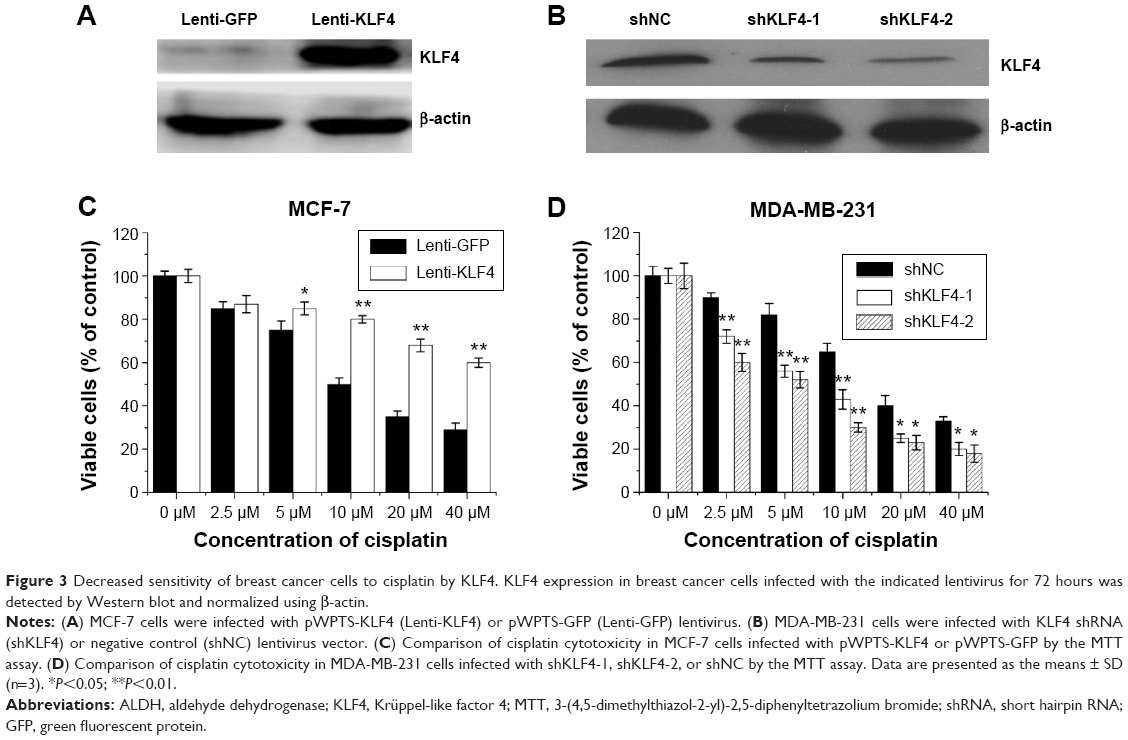 | Figure 3 Decreased sensitivity of breast cancer cells to cisplatin by KLF4. KLF4 expression in breast cancer cells infected with the indicated lentivirus for 72 hours was detected by Western blot and normalized using β-actin. |
Increased sensitivity of breast cancer cells to MCL by KLF4
KLF4 marks a phenotype that is highly resistant to cisplatin; however, to our surprise, MCF-7 cells infected with pWPTS-KLF4 lentivirus showed no apoptosis resistance toward MCL compared with controls (Figure 4C). Furthermore, overexpression of KLF4 in MCF-7 cells resulted in increased sensitivity to MCL treatment (Figure 4A). Conversely, KLF4 knockdown by lentivirus transduction was shown to attenuate the antiproliferative effects of MCL in MDA-MB-231 cells (Figure 4B). Therefore, flow cytometry with double staining of annexin V-PE/7-amino-actinomycin D (7-AAD) was performed to discriminate apoptotic and necrotic cell death. This analysis demonstrated that the cell apoptosis rate of MCF-7 cells overexpressing KLF4 was enhanced (Figure 4C), which indicated that MCF-7 cells with high KLF4 levels were more sensitive to MCL-induced cell apoptosis.
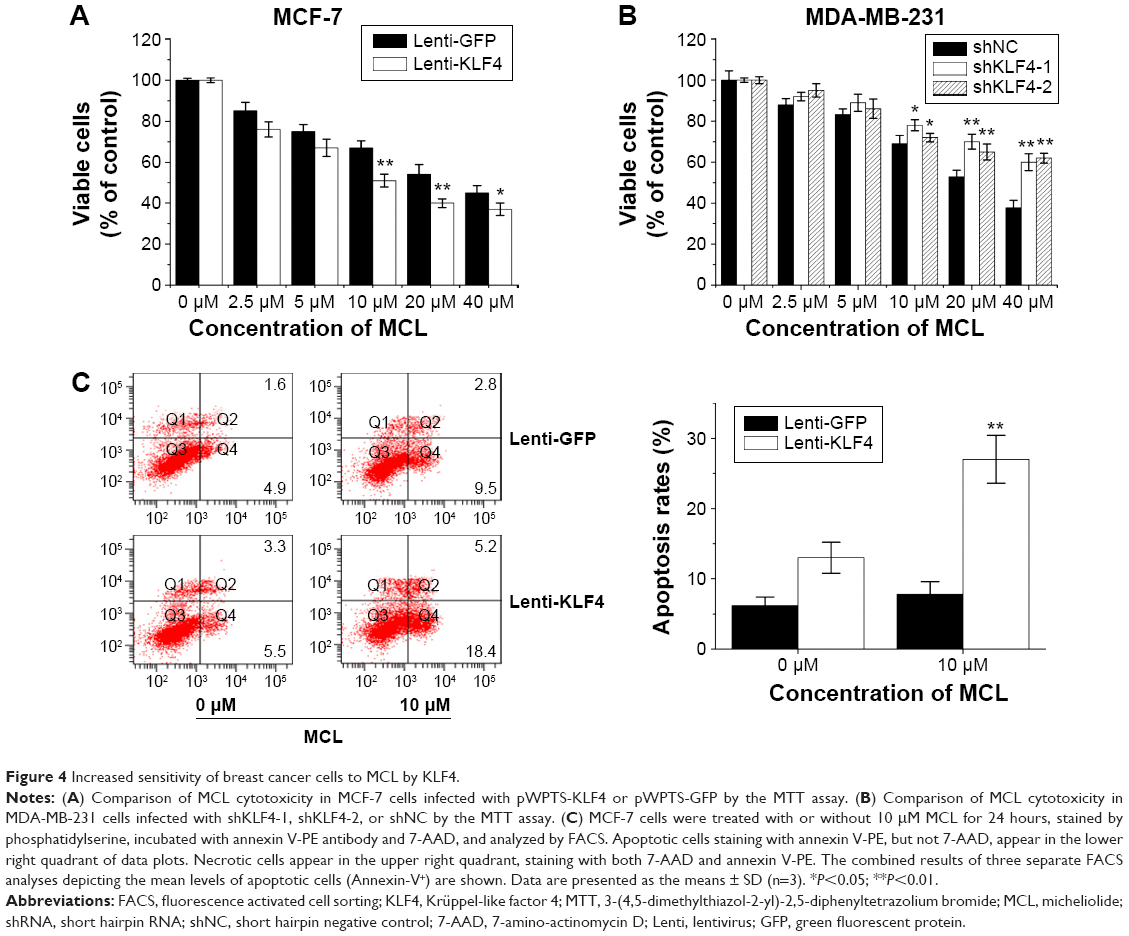 | Figure 4 Increased sensitivity of breast cancer cells to MCL by KLF4. |
MCL downregulates intracellular levels of GSH in MCF-7 cells
In our previous study, we found that the expression of KLF4 was positively correlated with the intracellular level of GSH in hepatocarcinoma.5 Similarly, the intracellular level of GSH was upregulated after KLF4 overexpression in breast cancer MCF-7 cells (Figure 5A), suggesting that GSH might contribute to KLF4-mediated cisplatin resistance. As shown in Figure 5B, MCL could downregulate the intracellular level of GSH in a concentration-dependent manner. Overexpression of KLF4 accompanied acquired cisplatin resistance in MCF-7 cells and augmented their sensitivity to MCL. Buthionine sulphoximine (BSO) is an inhibitor of gamma-glutamylcysteine synthetase (gamma-GCS) and, consequently lowers tissue GSH concentrations. Since BSO does not affect cytochrome P-450 or conjugating enzyme activity, its use as a specific depletor of tissue GSH in the investigation of mechanisms of xenobiotic-induced toxicities is preferable to the standard GSH-depleting agents as these have other enzymic effects.14 We next measured the cytotoxic effects in cells exposed to different concentrations of cisplatin in the presence or absence of BSO or MCL. The results showed that pre-incubation with BSO or MCL abolished the cellular resistance effects of KLF4 on cisplatin-induced cytotoxicity in MCF-7 cells (Figure 5C), and that the KLF4-induced cellular resistance to cisplatin in MCF-7 cells occurred in a GSH-dependent manner. Thus, these results suggest that MCL overcomes KLF4-mediated cisplatin resistance by downregulating GSH in MCF-7 cells.
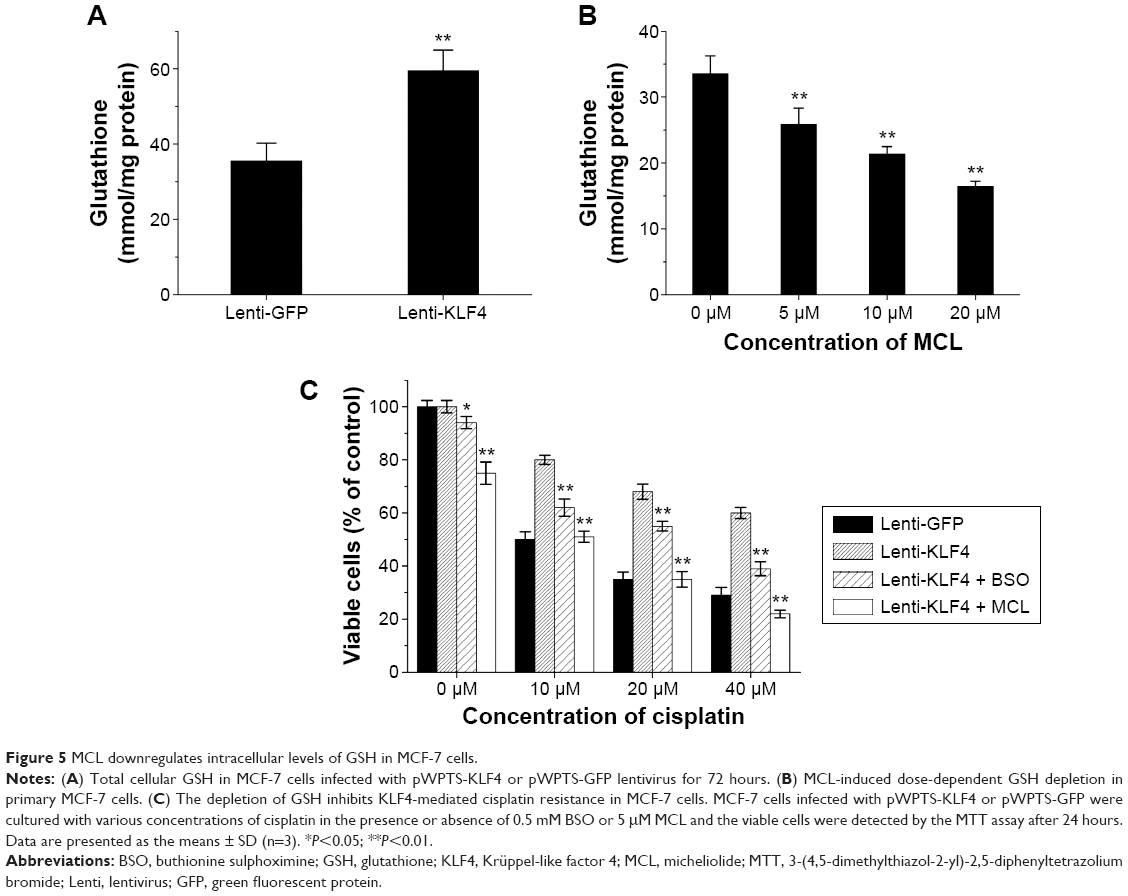 | Figure 5 MCL downregulates intracellular levels of GSH in MCF-7 cells. |
Discussion
Cisplatin is a widely used chemotherapeutic agent in advanced breast cancer, but many patients eventually relapse and acquired or de novo resistance is virtually universal.15 Therefore, the elucidation of the mechanisms of chemoresistance and the discovery of novel anticancer drugs are required to improve the overall survival in patients with chemoresistant metastatic breast cancer.16 Cisplatin is a well-known DNA-damaging agent, and the primary antineoplastic activity of cisplatin is generally believed to involve its interaction with chromosomal DNA and the subsequent induction of apoptosis.8 However, only 1% of intracellular cisplatin forms covalent bonds with nuclear DNA17 and cisplatin exerts prominent cytotoxic effects in enucleated cells (cytoplasts).18,19 Furthermore, cisplatin is poorly soluble in water, and it undergoes chemical reactions with other molecules once inside the cell. Initially, its chloride is replaced with water forming spontaneously in the cytoplasm. The aquated form of cisplatin is highly electrophilic and hence prone to form covalent bonds with a large panel of cysteine-containing peptides and polypeptides, including reduced GSH.20 While conjugation of cisplatin to GSH is the first step in the activation of cisplatin to a nephrotoxin, it renders cisplatin inactive as an antitumor drug. In addition, conjugation of cisplatin with GSH reduces the formation of interstrand and intrastrand platinum DNA adducts, resulting in the decreased toxicity of cisplatin in dividing cells.21 In our study, treatment of the MCF-7 cell line with low-dose cisplatin (10 μM) was sufficient to enrich the proportion of ALDH+ cells, to impact mammosphere formation, and to upregulate KLF4 expression; the cisplatin-induced enrichment of ALDH+ cells was found to be mediated through increased levels of KLF4. The results obtained from knockdown and overexpression experiments in breast cancer cell lines demonstrated that KLF4 is both necessary and sufficient to induce a cisplatin resistance phenotype. Thus, KLF4-overexpressing MCF-7 cell lines were considered feasible models for drug screening to overcome cisplatin resistance. To our surprise, the KLF4-overexpressing MCF-7 cell lines were much more sensitive to MCL treatment than were the controls. Accordingly, MCL could enhance the cisplatin sensitivity of MCF-7 cells both in vitro and in vivo. Furthermore, the content of GSH was found to be elevated in MCF-7 cells after overexpression of KLF4. Total cellular GSH content is an important determinant of cisplatin resistance, and the association between elevated cellular levels of GSH and cisplatin resistance has led to the suggestion that inactivation of cisplatin could occur through conjugation with GSH.22,23 KLF4-mediated resistance to cisplatin in MCF-7 cells was found to be completely abrogated by treatment with BSO, an inhibitor of GSH synthesis. These findings led us to hypothesize that the cisplatin sensitivity of MCF-7 cells would be responsive to inhibition of GSH. To test this premise, we identified MCL as a compound that induced GSH depletion and severe cell death in KLF4-overexpressing MCF-7 cells exposed to cisplatin; therefore, MCL-mediated direct depletion of GSH represented a major mechanism in reversing KLF4-induced cisplatin resistance in MCF-7 cells.
Current knowledge about the evolution of the breast carcinogenic process must include CSCs, which are essential to understand tumor occurrence, recurrence, and chemoresistance.24 Recent evidence suggests that an altered redox status and acquired aberrant GSH metabolism are implicated in the etiology of CSCs and that CSCs take advantage of the aberrant redox system and spontaneously proliferate.6,25 As a novel compound, MCL can effectively overcome breast cancer stem-like KLF4-mediated cisplatin resistance. This observation is important because it indicates that suppression of GSH is sufficient to augment the activities of cisplatin, suggesting that MCL might be useful as an adjuvant to cisplatin.
Conclusion
These findings demonstrate that KLF4 expression enhanced cisplatin resistance in breast cancer cells by increasing the levels of intracellular GSH. Furthermore, MCL, which induces GSH depletion, can effectively overcome KLF4-mediated cisplatin resistance in breast cancer cells.
Acknowledgments
This work was supported by grants from the National Science and Technology Pillar Program (2015BAI12B15), the National Nature Science Foundation of China (81302080), the Anticancer Key Technologies R&D Program of Tianjin (12ZCDZSY16200), the Research Fund for the Doctoral Program of Higher Education of China (20131202120003), the Natural Science Foundation of Tianjin (14JCQNJC11100), and the Foundation of Tianjin Medical University (2013KYQ06).
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in either drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agreement to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang P, Andrianakos R, Yang Y, Liu C, Lu W. Kruppel-like factor 4 (klf4) prevents embryonic stem (es) cell differentiation by regulating nanog gene expression. J Biol Chem. 2010;285:9180–9189. | ||
Pandya AY, Talley LI, Frost AR, et al. Nuclear localization of klf4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10:2709–2719. | ||
Yu F, Li J, Chen H, et al. Kruppel-like factor 4 (klf4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. | ||
Dong MJ, Wang LB, Jiang ZN, Jin M, Hu WX, Shen JG. The transcription factor klf4 as an independent predictive marker for pathologic complete remission in breast cancer neoadjuvant chemotherapy: a case-control study. Onco Targets Ther. 2014;7:1963–1969. | ||
Jia Y, Zhang W, Liu, H, Peng L, Yang Z, Lou J. Inhibition of glutathione synthesis reverses kruppel-like factor 4-mediated cisplatin resistance. Cancer Chemother Pharmacol. 2012;69:377–385. | ||
Pei S, Minhajuddin M, Callahan KP, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288:33542–33558. | ||
Nagano O, Okazaki S, Saya H. Redox regulation in stem-like cancer cells by cd44 variant isoforms. Oncogene. 2013;32:5191–5198. | ||
Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. | ||
Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. | ||
Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. | ||
Zhang J, Wang X, Cui W, et al. Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nat Commun. 2013;4:2157. | ||
Yde CW, Issinger OG. Enhancing cisplatin sensitivity in mcf-7 human breast cancer cells by down-regulation of bcl-2 and cyclin d1. Int J Oncol. 2006;29:1397–1404. | ||
Ginestier C, Hur MH, Charafe-Jauffret E, et al. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. | ||
Drew R, Miners JO. The effects of buthionine sulphoximine (bso) on glutathione depletion and xenobiotic biotransformation. Biochem Pharmacol. 1984;33:2989–2994. | ||
Cobleigh MA. Other options in the treatment of advanced breast cancer. Semin Oncol. 2011;38(Suppl 2):S11–S16. | ||
Shajahan-Haq AN, Cheema MS, Clarke R. Application of metabolomics in drug resistant breast cancer research. Metabolites. 2015;5:100–118. | ||
Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–663. | ||
Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res. 2006;12:5817–5825. | ||
Yu F, Megyesi J, Price PM. Cytoplasmic initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol. 2008;295:F44–F52. | ||
Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. | ||
Townsend DM, Deng M, Zhang L, Lapus M.G, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol. 2003;14:1–10. | ||
Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci U S A. 1992;89:3070–3074. | ||
Zhang K, Chew M, Yang EB, Wong KP, Mack P. Modulation of cisplatin cytotoxicity and cisplatin-induced DNA cross-links in hepg2 cells by regulation of glutathione-related mechanisms. Mol Pharmacol. 2001;59:837–843. | ||
Nguyen NP, Almeida FS, Chi A, et al. Molecular biology of breast cancer stem cells: potential clinical applications. Cancer Treat Rev. 2010;36:485–491. | ||
Ogasawara MA, Zhang H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxid Redox Signal. 2009;11:1107–1122. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.