")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Insulin induces drug resistance in melanoma through activation of the PI3K/Akt pathway
Authors Chi M, Ye Y, Zhang XD , Chen J
Received 26 August 2013
Accepted for publication 5 November 2013
Published 17 February 2014 Volume 2014:8 Pages 255—262
DOI https://doi.org/10.2147/DDDT.S53568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Mengna Chi,1 Yan Ye,1 Xu Dong Zhang,1 Jiezhong Chen2,3
1School of Medicine and Public Health, The University of Newcastle, Newcastle, NSW, Australia; 2School of Biomedical Sciences, The University of Queensland, Brisbane, QLD, Australia; 3Faculty of Science, Medicine and Health, The University of Wollongong, Wollongong, NSW, Australia
Introduction: There is currently no curative treatment for melanoma once the disease spreads beyond the original site. Although activation of the PI3K/Akt pathway resulting from genetic mutations and epigenetic deregulation of its major regulators is known to cause resistance of melanoma to therapeutic agents, including the conventional chemotherapeutic drug dacarbazine and the Food and Drug Administration-approved mutant BRAF inhibitors vemurafenib and dabrafenib, the role of extracellular stimuli of the pathway, such as insulin, in drug resistance of melanoma remains less understood.
Objective: To investigate the effect of insulin on the response of melanoma cells to dacarbazine, and in particular, the effect of insulin on the response of melanoma cells carrying the BRAFV600E mutation to mutant BRAF inhibitors. An additional aim was to define the role of the PI3K/Akt pathway in the insulin-triggered drug resistance.
Methods: The effect of insulin on cytotoxicity induced by dacarbazine or the mutant BRAF inhibitor PLX4720 was tested by pre-incubation of melanoma cells with insulin. Cytotoxicity was determined by the MTS assay. The role of the PI3K/Akt pathway in the insulin-triggered drug resistance was examined using the PI3K inhibitor LY294002 and the PI3K and mammalian target of rapamycin dual inhibitor BEZ-235. Activation of the PI3K/Akt pathway was monitored by Western blot analysis of phosphorylated levels of Akt.
Results: Recombinant insulin attenuated dacarbazine-induced cytotoxicity in both wild-type BRAF and BRAFV600E melanoma cells, whereas it also reduced killing of BRAFV600E melanoma cells by PLX4720. Nevertheless, the protective effect of insulin was abolished by the PI3K and mTOR dual inhibitor BEZ-235 or the PI3K inhibitor LY294002.
Conclusion: Insulin attenuates the therapeutic efficacy of dacarbazine and PLX4720 in melanoma cells, which is mediated by activation of the PI3K/Akt pathway and can be overcome by PI3K inhibitors.
Keywords: insulin, PI3K/Akt, melanoma, drug resistance, DTIC, BRAF inhibitors
Introduction
Melanoma is one of the most aggressive cancers. Although early stages of melanoma can be removed by surgery with high patient survival rates, metastatic melanoma has poor prognosis. Despite recent progress in the treatment of late-stage melanoma, and in particular, the advance in development of novel targeted therapy and immunotherapy approaches, the overall outcome of patients with metastatic melanoma remains poor.1 The median survival period of these patients is about 6 to 10 months, and the 5-year survival rate is lower than 20%.2,3 Therefore, it is of paramount importance to improve the therapeutic efficacy of treatments for metastatic melanoma. This could be achieved either by improving already existing Food and Drug Administration-approved drugs or by developing novel therapeutic agents.
Dacarbazine (DTIC) is the most commonly used chemotherapeutic drug in the treatment of metastatic melanoma.4 However, only 5%–10% of patients benefit from administration of DTIC.5 The mechanisms responsible for resistance of melanoma cells to DTIC remain undefined, but combinations of DTIC with other agents have been attempted to increase the therapeutic efficacy. A Phase III clinical trial showed that DTIC in combination with cisplatin or vindesine increased the response rate to 20%–40%.6 However, disease-free and overall survival rates were not increased.7 The combination of DTIC with interleukin-2 or tumor-necrosis-factor-α improved the response rate, but adverse effects were also increased.8
Cancer-targeted therapy is based on an understanding of the roles of key molecules in the cancer pathogenesis. Activating BRAF mutations that drive melanoma cell survival and proliferation are found in about 60% of melanomas. The most common mutation in melanoma is a glutamic acid substitution for valine at position 600 (BRAFV600E).9,10 Targeting mutant BRAF using small molecule inhibitors, such as vemurafenib and dabrafenib has achieved unprecedented responses in metastatic melanoma patients.11–13 However, complete remission is rare and a proportion of mutant BRAF melanomas are less responsive to the inhibitors, indicative of inherent resistance.11,14–16 Moreover, the durations of responses are commonly limited, with most patients relapsing within 1 year, which is indicative of development of acquired drug resistance.11,14–16 Multiple mechanisms have been shown to contribute to the resistance of mutant BRAF melanomas to BRAF inhibitors.11,14–16 These include those mechanisms leading to insufficient inhibition of RAF/MEK/ERK signaling and those promoting melanoma cell survival and proliferation alternative to the RAF/MEK/ERK pathway, such as increased activation of the PI3K/Akt or NF-κB pathways.17–25 Indeed, combinations of RAF inhibitors and inhibitors of MEK such as trametinib to further inhibit MEK/ERK signaling have yielded promising results in clinical trials.26–28 Co-targeting the PI3K/Akt and RAF/MEK/ERK pathways is also being evaluated in early clinical studies.23,29
Insulin is known to activate the PI3K/Akt pathway and thus increases carcinogenesis.30,31 Besides gene mutations of the major components of the pathway, extracellular stimuli such as insulin and insulin-like growth factor 1 (IGF-1) also enhance PI3K/Akt signaling and cause drug resistance. Indeed, in many types of cancer cells, insulin induces resistance to chemotherapeutic drugs and may contribute to poor prognosis, particularly in patients with obesity and type II diabetes.32,33 However, the role of insulin in melanoma cell resistance to chemotherapy and targeted therapy remains less understood. Here we show that insulin attenuates the therapeutic efficacy of DTIC and the BRAF inhibitor PLX4720 in melanoma cells, and that this is mediated by activation of the PI3K/Akt pathway and can be overcome by PI3K inhibitors and dual inhibitors of PI3K and mammalian target of rapamycin (mTOR).
Materials and methods
Cell culture and reagents
The mouse melanoma cell line B16 and the human melanoma cell line Mel-RMu described previously were cultured in Dulbecco’s Modified Eagle’s Medium containing 5% fetal calf serum.34 Antibodies against Akt and phospho-Akt (Ser473) were purchased from Cell Signaling Technology (Danvers, MA, USA). The PI3K inhibitor LY294002 was from Calbiochem (Merck KGaA, Darmstadt, Germany). The dual inhibitor PI3K and mTOR, BEZ-235, was from Selleckchem (Houston, TX, USA). The antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were from Ambion (Life Technologies, Carlsbad, CA, USA). The BRAF inhibitor PLX4720 was from Selleckchem. Dacarbazine was purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell viability
Cell viability was determined by MTS assay using Celltiter 96 aqueous one solution cell proliferation assay kit according to the manufacturer’s instructions (Promega Corporation, Madison, WI, USA) as described previously.35 In brief, cells were seeded at 5,000 cells/well onto flat-bottomed 96-well culture plates and allowed to grow for 24 hours followed by the desired treatment duration. Cells were then labeled with the VisionBlueTM reagent and detected by Synergy 2TM multi-detection microplate reader (Biotek, Winooski, VT, USA).
Immunoblotting
Immunoblotting was carried out as described previously.35 Labeled bands were detected by Luminata Crescendo Western horseradish peroxidase substrate (EMD Millipore, Billerica, MA, USA) and images were captured and the intensity of the bands was quantitated using an ImageReader LAS-4000 (Fujifilm Corporation, Tokyo, Japan).
Statistical analysis
Statistical analysis was performed using JMP Statistics Made VisualTM software (SAS Institute, Cary, NC, USA). Student’s t-test was used to assess differences in the values of different experimental groups. P-values less than 0.05 were considered statistically significant.
Results
Insulin protects melanoma cells against DTIC-mediated cytotoxicity
We examined the cytotoxic effect of DTIC on the mouse melanoma cell line B16 that harbored wild-type BRAF and the human melanoma cell line Mel-RMu that carried BRAFV600E by treating the cells with the drug at increasing concentrations for 24 hours.36,37 As shown in Figure 1, DTIC reduced viability of B16 and Mel-RMu cells in a dose-dependent manner irrespective of their BRAF mutational status. Since DTIC at 25 μg/mL inhibited viability of both B16 and Mel-RMu cells by approximately 50% (Figure 1). This concentration was chosen for assays hereafter.
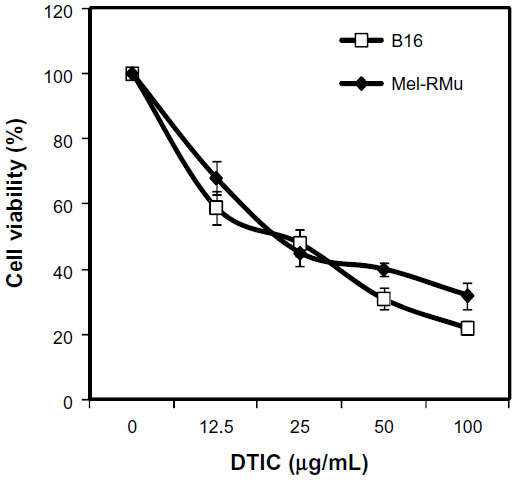 | Figure 1 Cytotoxicity of DTIC towards melanoma cells. |
To examine whether insulin protects melanoma cells from cytotoxicity induced by DTIC, B16 and Mel-RMu cells were pre-treated with insulin at a range of concentrations for 15 minutes before the addition of DTIC at 25 μg/mL for a further 24 hours. As shown in Figure 2, even when used at 250 nM, insulin significantly protected Mel-RMu and, to a lesser extent, B16 cells from DTIC-mediated cytotoxicity. The survival rates of Mel-RMu and B16 cells were increased by 40% and 25%, respectively (Figure 2). Notably, insulin at increased concentrations did not afford any further protection against DTIC. This may be due to negative cooperativity of binding of insulin to its receptor.38 Regardless, these results suggest that circulating insulin in melanoma patients, particularly melanoma patients with obesity, may attenuate the therapeutic efficacy of chemotherapeutic drugs.
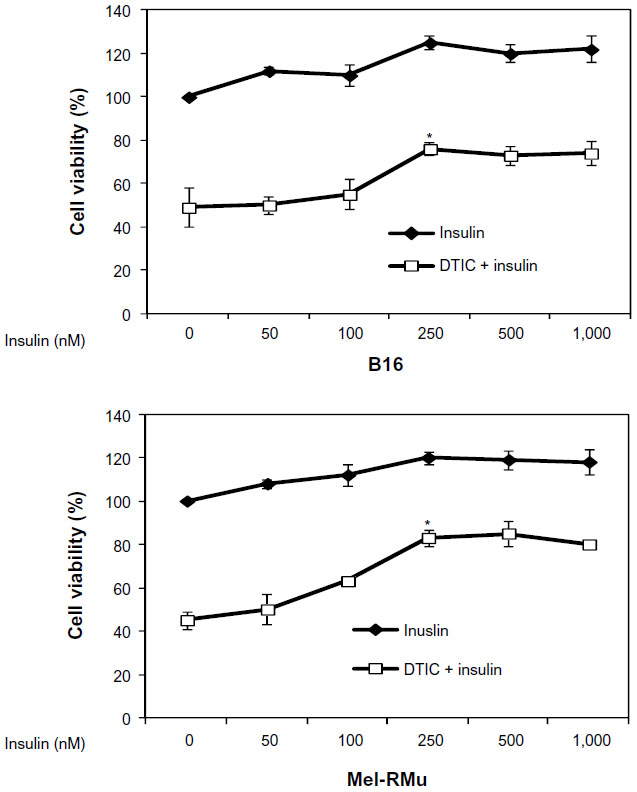 | Figure 2 Insulin protects melanoma cells against DTIC. |
Insulin protects BRAFV600E melanoma cells from cytotoxicity induced by the mutant BRAF inhibitor PLX4720
Since Mel-RMu cells are known to harbor BRAFV600E and are sensitive to the mutant BRAF inhibitor PLX4720,24 we examined whether insulin similarly protects the cells from PLX4720-mediated cytotoxicity. B16 mouse melanoma cells that carry wild-type BRAF and are not sensitive to mutant BRAF inhibitors were excluded from studies using PLX4720.39 Indeed, insulin at 250 nM significantly inhibited reduction in viability of Mel-RMu cells induced by PLX4720 (Figure 3).
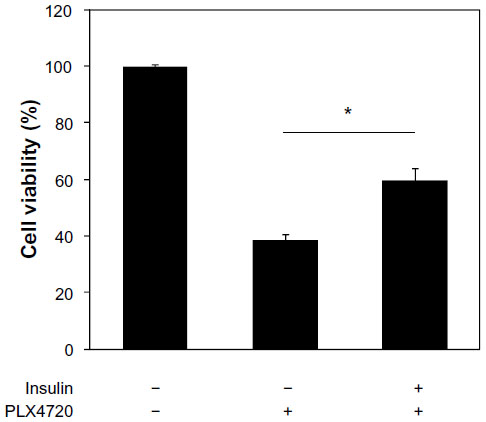 | Figure 3 Insulin protects BRAFV600E melanoma cells against the BRAF inhibitor PLX4720. |
Insulin activates the PI3K/Akt signalling pathway in melanoma cell lines
Aberrant activation of the PI3K/Akt pathway is known to confer resistance of melanoma cells to therapeutic drugs.30,31 We examined whether exposure of melanoma cells to insulin results in activation of PI3K/Akt signaling. As anticipated, exposure to insulin increased the levels of phosphorylated Akt in both B16 and Mel-RMu cells (Figure 4), indicating increased activation of the PI3K/Akt pathway.
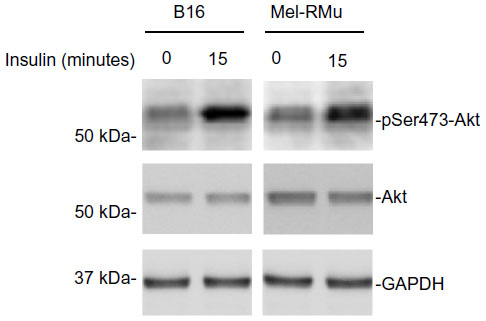 | Figure 4 Insulin activates the PI3K/Akt in melanoma cells. |
Inhibition of the PI3K/Akt pathway reverses protection of melanoma cells against DTIC and/or PLX4720 by insulin
To confirm the role of the PI3K/Akt pathway in insulin-mediated protection of melanoma cells from therapeutic drugs, we pre-treated B16 and Mel-RMu cells with the PI3K and mTOR dual inhibitor BEZ-235 before the addition of insulin (250 nM) followed by DTIC. While BEZ-235 abolished the increase in activation of Akt induced by insulin in both B16 and Mel-RMu cells (Figure 5A), it also diminished insulin-mediated protection against cytotoxicity induced by DTIC (Figure 5B). Similarly, BEZ-235 also significantly inhibited protection of Mel-RMu cells against PLX4720 by insulin (Figure 5C). Of note, BEZ-235 enhanced cytotoxicity triggered by PLX4720 alone in Mel-RMu cells (Figure 5C), consistent with previous reports that inhibition of the PI3K/Akt pathway sensitizes mutant BRAF melanoma cells to BRAF inhibitors.40,41
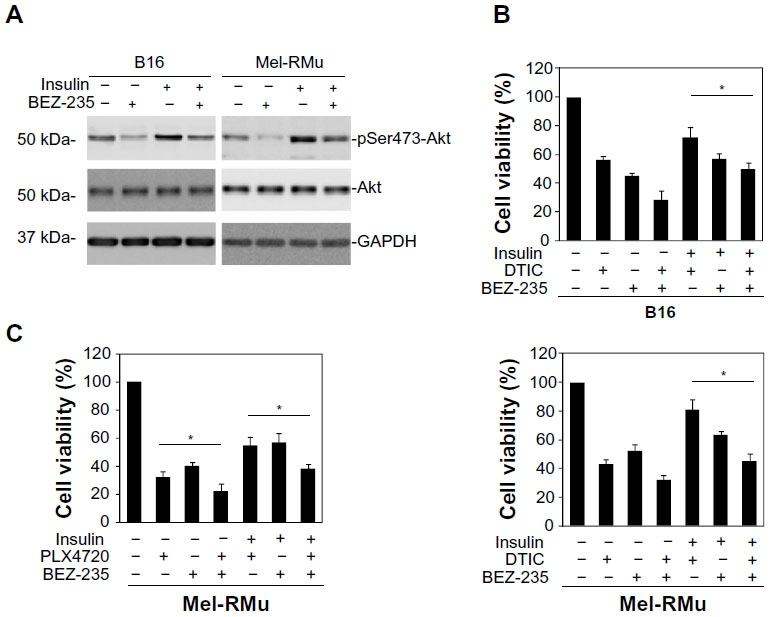 | Figure 5 The PI3K and mTOR dual inhibitor BEZ-235 reverses protection of melanoma cells against DTIC and/or PLX4720 by insulin. |
We also examined whether the PI3K inhibitor LY294002 similarly reverses protection of melanoma cells against therapeutic drugs by insulin. As shown in Figure 6A, LY294002 abolished the insulin-triggered increase in activation of Akt in B16 and Mel-RMu cells. Moreover, similar to BEZ-235, it also significantly inhibited insulin-mediated protection of B16 and Mel-RMu cells from DTIC, and protection of Mel-RMu cells from PLX4720 (Figure 6B and C). Together, these results suggest that activation of the PI3K/Akt pathways plays a predominant role in protection of melanoma cells from therapeutic drugs by insulin.
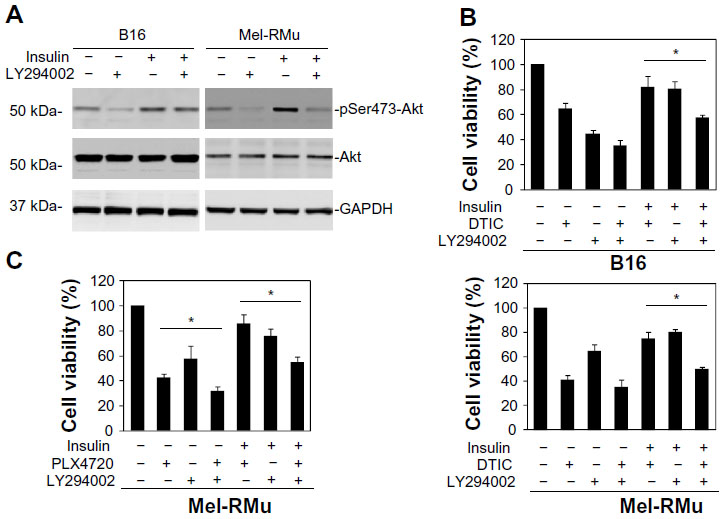 | Figure 6 The PI3K inhibitor LY294002 reverses protection of melanoma cells against DTIC and/or PLX4720 by insulin. |
Discussion
Resistance of melanoma cells to therapeutic drugs is a major obstacle in the quest for curative treatment of melanoma. The response rate to the conventional chemotherapeutic drug DTIC is only 5%–10%,5 whereas approximately 50%–60% of patients with mutant BRAF metastatic melanomas initially respond to the BRAF inhibitor vemurafenib or dabrafenib.11,14–16 Nevertheless, durations of responses to BRAF inhibitors are commonly limited, with most patients relapsing within 1 year.12,15,16 One of the mechanisms responsible for resistance of melanoma cells to therapeutic drugs is aberrant activation of the PI3K/Akt pathway due to genetic mutations and/or epigenetic deregulation of its major regulators.42,43 However, the role of extracellular stimuli that can activate the pathway in drug-resistant melanoma cells remains less well understood. In this report, we demonstrated that the addition of insulin enhanced resistance of cultured melanoma cells to DTIC. In addition, we showed that insulin also protected mutant BRAF melanoma cells from the BRAF inhibitor PLX4720. These results suggest that elevated levels of circulating insulin in melanoma patients, such as those with obesity and type II diabetes, may compromise their responses to conventional chemotherapeutic drugs and agents targeting pro-survival pathways, thus leading to poor prognosis.
Our study showed that insulin increased activation of the PI3K/Akt pathway, and that inhibition of the pathway reversed insulin-mediated protection against DTIC in both wild-type BRAF B16 cells and BRAFV600E Mel-RMu cells. In addition, the results also demonstrated that inhibition of cytotoxicity of PLX4720 by insulin in Mel-RMu cells were also abolished when PI3K/Akt signaling was blocked. These results indicate that activation of the PI3K/Akt pathway plays an essential role in drug resistance of melanoma cells induced by insulin. The PI3K/Akt pathway is a well-known pro-survival pathway in many cancers.30,31 The pathway is activated by various growth factors and regulates a broad range of targets that regulate cell survival, proliferation, and resistance to therapeutic agents.31 Our results showing that insulin activates the PI3K/Akt pathway leading to drug resistance reinforce the importance of extracellular stimuli of the pathway in protection of melanoma cells against treatment. Insulin is known to bind to insulin receptors, IGF-1 receptors, and insulin/IGF-1 hybrid receptors to activate PI3K/Akt.40 In addition, it can inhibit the production of IGF-binding proteins and therefore increase blood levels of IGF-1, a stronger anti-apoptotic molecule than insulin. Indeed, blood levels of IGF-1 have been reported to positively associate with the incidence of melanoma while blood levels of IGF-binding proteins are inversely associated.23 IGF-1 has been demonstrated to increase survival and growth of melanoma cells via activation of MAPK and PI3K/Akt pathways.44 It is highly possible that IGF-1 can also cause drug resistance to chemotherapeutic agents in melanoma cells. In addition to insulin, a number of other growth factors that may exist in a melanoma microenvironment, such as fibroblast growth factor, hepatocyte growth factor, and epithelial growth factor, are conceivably involved in drug resistance of melanoma cells, as they can similarly stimulate PI3K/Akt signaling and other survival pathways such as the MEK/ERK pathway.
DTIC is a standard drug that is commonly used in the treatment of metastatic melanoma. Although the initial response rate is low, the drug is not out of date.4,5 Our results showing that activation of the PI3K/Akt pathway by insulin protects melanoma cells from DTIC could partially explain the low response rate to DTIC, and suggest that the combinatorial approaches with inhibitors of the PI3K/Akt pathway and DTIC may increase the therapeutic efficacy. Combinations of DTIC with other agents have not been satisfactory.6,8
It is of note that the PI3K inhibitor LY294002 and the PI3K and mTOR dual inhibitor BEZ-235 decreased the effect of insulin on protection of melanoma cells against therapeutic drugs to a similar extent, suggesting that the protective effect of insulin is mainly mediated by PI3K, although mTOR may also play a role. Various inhibitors of the PI3K/Akt pathway and dual inhibitors of PI3K and mTOR are in clinical and/or preclinical studies, including BEZ-235.45 Our results support combinations of BEZ-235 and other PI3K inhibitors with clinically available agents for further evaluation. One of the suitable conditions for such a regime is in the case of patients with elevated serum levels of insulin, such as those with obesity and type II diabetes.
In summary, we have provided evidence that insulin protects melanoma cells from the chemotherapeutic drug DTIC and BRAF inhibitors. In addition, we have shown that insulin-induced drug resistance is primarily due to activation of the PI3K/Akt pathway. Our results indicate that combinations of inhibitors of the PI3K/Akt pathway and DTIC or BRAF inhibitors may be useful to improve the therapeutic efficacy in the treatment of melanoma, in particular, in patients with obesity and type II diabetes who have elevated serum insulin levels.
Disclosure
The authors report no conflicts of interest in this work.
References
Boyle GM. Therapy for metastatic melanoma: an overview and update. Expert Rev Anticancer Ther. 2011;11(5):725–737. | |
Falkson CI, Ibrahim J, Kirkwood JM, Coates AS, Atkins MB, Blum RH. Phase III trial of dacarbazine versus dacarbazine with interferon alpha-2b versus dacarbazine with tamoxifen versus dacarbazine with interferon alpha-2b and tamoxifen in patients with metastatic malignant melanoma: an Eastern Cooperative Oncology Group study. J Clin Oncol. 1998;16(5):1743–1751. | |
Atkins MB, Hsu J, Lee S, et al; Eastern Cooperative Oncology Group. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2008;26(35):5748–5754. | |
Eigentler TK, Caroli UM, Radny P, Garbe C. Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol. 2003;4(12):748–759. | |
Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. | |
Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27(17):2823–2830. | |
Pectasides D, Yianniotis H, Alevizakos N, et al. Treatment of metastatic malignant melanoma with dacarbazine, vindesine and cisplatin. Br J Cancer. 1989;60(4):627–629. | |
Hamm C, Verma S, Petrella T, Bak K, Charette M; Melanoma Disease Site Group of Cancer Care Ontario’s Program in Evidence-based Care. Biochemotherapy for the treatment of metastatic malignant melanoma: a systematic review. Cancer Treat Rev. 2008;34(2):145–156. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Smalley KS. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J Invest Dermatol. 2010;130(1):28–37. | |
Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAFV600E mutation. N Engl J Med. 2011;364(26):2507–2516. | |
Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19(1):3–4. | |
Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8(7):426–433. | |
Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11(11):873–886. | |
Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132(3 Pt 2):854–863. | |
Smalley KS, Sondak VK. Melanoma – an unlikely poster child for personalized cancer therapy. N Engl J Med. 2010;363(9):876–878. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | |
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | |
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. | |
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | |
Paraiso KH, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. | |
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | |
Jiang CC, Lai F, Thorne RF, et al. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res. 2011;17(4):721–730. | |
Madonna G, Ullman CD, Gentilcore G, Palmieri G, Ascierto PA. NF-κB as potential target in the treatment of melanoma. J Transl Med. 2012;10:53. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–789. | |
Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773–781. | |
Shimizu T, Tolcher AW, Papadopoulos KP, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–2325. | |
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159–169. | |
Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18(1):40–50. | |
Chen J, Katsifis A, Hu C, Huang XF. Insulin decreases therapeutic efficacy in colon cancer cell line HT29 via the activation of the PI3K/Akt pathway. Curr Drug Discov Technol. 2011;8(2):119–125. | |
Chen J, Huang XF, Qiao L, Katsifis A. Insulin caused drug resistance to oxaliplatin in colon cancer cell HT29. J Gastrointest Oncol. 2011;2(1):27–33. | |
Gillespie S, Zhang XD, Hersey P. Variable expression of protein kinase C epsilon in human melanoma cells regulates sensitivity to TRAIL-induced apoptosis. Mol Cancer Ther. 2005;4(4):668–676. | |
Tay KH, Jin L, Tseng HY, et al. Suppression of PP2A is critical for protection of melanoma cells upon endoplasmic reticulum stress. Cell Death Dis. 2012;3:e337. | |
Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. | |
York RM, Foltz AT. Bleomycin, vincristine, lomustine, and DTIC chemotherapy for metastatic melanoma. Cancer. 1988;61(11):2183–2186. | |
Taylor SI, Leventhal S. Defect in cooperativity in insulin receptors from a patient with a congenital form of extreme insulin resistance. J Clin Invest. 1983;71(6):1676–1685. | |
Di Virgilio S, Hellmann KP, Robberecht P, et al. Multiple differences between wild-type B16 melanoma cells and a wheat germ agglutinin resistant clone. Anticancer Res. 1991;11(5):1815–1821. | |
Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3(2):158–167. | |
Wroblewski D, Mijatov B, Mohana-Kumaran N, et al. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis. 2013;34(2):237–247. | |
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | |
Chen J, Shao R, Zhang XD, Chen C. Applications of nanotechnology for melanoma treatment, diagnosis, and theranostics. Int J Nanomedicine. 2013;8:2677–2688. | |
Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and β-catenin pathways. Cancer Res. 2001;61(19):7318–7324. | |
Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68(19):8022–8030. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.