")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Insights into the genetic basis of systemic sclerosis: immunity in human disease and in mouse models
Received 16 May 2014
Accepted for publication 5 July 2014
Published 26 September 2014 Volume 2014:4 Pages 143—151
DOI https://doi.org/10.2147/AGG.S46813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Minghua Wu, Maureen D Mayes
Division of Rheumatology and Clinical Immunogenetics, Department of Internal Medicine, University of Texas Medical School at Houston, Houston, TX, USA
Abstract: Systemic sclerosis (SSc; scleroderma) is a chronic, multisystem autoimmune disease characterized by vasculopathy, fibrosis, and autoantibodies. In the past decade, great efforts have been made to investigate genetic susceptibility for SSc. To date, over 20 gene loci have been identified as risk factors for SSc in large genome-wide association studies and confirmed by independent replication studies. However, the biological relevance of these genetic associations is still largely unknown. Exploring the mechanism behind these risk loci is essential to better understand disease pathogenesis and to identify novel therapeutic targets. Mouse model studies including knockout, knockin and knockdown of these genes can advance our understanding of pathogenic cellular and molecular mechanisms in human disease. Although such mouse model systems do not exactly correspond to human disease, they can provide insight into pathological mechanisms that influence disease pathways. In this review, we discuss recent findings regarding the genetic basis of SSc in the setting of genetic manipulation of these pathways in murine models.
Keywords: GWAS, Immunochip study, type I interferon pathway, genetic mutation animal models
Introduction
Systemic sclerosis (scleroderma, SSc) is a chronic, multisystem autoimmune disease characterized by vasculopathy, fibrosis, and autoimmunity.1 At present, the etiology of SSc is unknown and disease-related mortality remains high. Reported prevalence and incidence estimates of SSc vary widely (prevalence estimates range from 31–659/million population and incidence rates range from 4–23/million/year) according to geographic location, ethnicity, and methods of case ascertainment.2 The interplay among genetic background, environmental factors, and autoimmunity as they relate to SSc pathogenesis is unclear. A comprehensive understanding of the genetic and molecular basis of SSc is crucial in order to develop appropriately targeted therapies.
In this review, we summarize our current knowledge about the genetic basis of SSc susceptibility identified in genome-wide association studies (GWASs) and related studies, and we present recent evidence from mouse models of SSc. We also discuss the consistency and discrepancy between findings derived from human and animal studies.
Heritability of SSc
In spite of recent advances in identifying genetic risk loci for SSc, only a small proportion of heritability has been explained in this complex autoimmune disease. Family studies have demonstrated that the risk of SSc is increased among first-degree relatives of patients, compared to the general population. In a study of 703 families in the United States, including 11 multiplex SSc families, familial SSc occurred significantly more frequently in siblings and first-degree relatives of SSc cases compared to in the population as a whole, with a relative risk (RR) of 15.3 A family history of SSc is the strongest risk factor identified to date, although the absolute risk is low. The largest SSc–twin study published thus far, with 42 twin pairs including 24 monozygotic pairs, failed to demonstrate a higher disease concordance in the monozygotic versus the dizygotic twins. However, concordance for the presence of antinuclear antibodies was significantly higher in the monozygotic twin pairs (90%) than in the dizygotic pairs (40%),4 suggesting that there was a shared genetic susceptibility to autoimmunity even among the unaffected co-twins. Further study in the same cohort showed a higher level of concordance among monozygotic twins regarding the gene expression profile of cultured dermal fibroblasts,4 demonstrating that a stronger genetic predisposition to SSc is apparent at the molecular level in skin fibroblasts in the monozygotic twins. A population-based study utilizing the Utah Population Database identified a significantly increased relative risk for Raynaud’s phenomenon (RR =6.38, 95% confidence interval [CI] =3.44–11.83) among first-degree relatives of SSc subjects compared to non-SSc families, as well as increased risk of other autoimmune diseases including systemic lupus erythematosus (SLE), Sjögren’s syndrome, dermatomyositis, rheumatoid arthritis, and interstitial lung disease.5 Taken together, these studies demonstrate the presence of a genetic predisposition to autoimmunity in general and SSc in particular.
Candidate gene studies, GWAS, and Immunochip analysis in SSc
The contribution of different genetic factors to SSc susceptibility in general and to its clinical and autoantibody subsets is now widely accepted. Multiple candidate gene studies in relatively small SSc cohorts6,7 have reported susceptibility loci for SSc. Unlike a candidate gene approach in which single nucleotide polymorphisms (SNPs) in a small number of genes are tested for association between cases and controls, a GWAS provides an unbiased view of SNP variants across the human genome. The Immunochip array is a custom SNP genotyping array with high density mapping of autoimmune disease (AID)-associated loci. The first large GWAS report of Radstake et al in 2010, followed by the GWAS report of Allanore et al in 2011, both with independent discovery and validation cohorts, identified the strongest association with SSc to reside in the major histocompatibility complex (MHC) class II region.8–10 In the human leukocyte antigen (HLA) region, the HLA-DQB1 locus was highly associated with the anticentromere antibody (ACA)-positive subgroup of SSc, and the HLA-DPB1 locus was associated with antitopoisomerase antibody (ATA)-positive SSc. In the MHC class III region, the NOTCH4 gene was highly associated with both ACA-positive and ATA-positive subgroups.9 Susceptibility alleles of the MHC region were well established by GWASs and the Immunochip study and are summarized in Table 1. Additional associated genes in the MHC region include the non-HLA genes TAP2, MICA, and MICB, which encode membrane proteins involved in antigen presentation and immune cell activation. These studies also emphasized the contribution of differential genetic components in the development of subphenotypes of SSc.
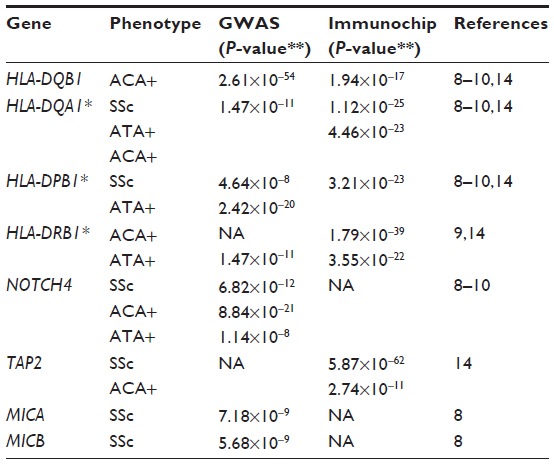 | Table 1 Established susceptibility alleles in MHC region from SSc GWASs and/or Immunochip study |
The GWAS and the subsequent Immunochip study also identified and firmly established the role of multiple SSc-susceptibility genes outside the MHC region, including CD247, interferon regulatory factors (IRFs) 5 and 8 (intergenic region TNPO3-IRF5 and IRF8), and signal transducer and activator of transcription 4 (STAT4). In addition, these and subsequent studies confirmed the association of previously reported genes, including TNIP1, TNFAIP3, IL12RB2, CSK, TNFSF4, BANK1, BLK, PTPN22, and PPARG, as SSc susceptibility loci (Table 2).6,10–13 Utilizing the Immunochip array, three new SSc risk loci were identified in a large multiple cohort study, including DNASE1L3 at 3p14, the SCHIP1-IL12A locus at 3q25, and ATG5 at 6q21, as well as a suggested association of the TREHDDX6 locus at 11q23.14 The associations of SSc risk loci STAT4, IRF5, and PXK genes, which had been previously reported in GWASs, were again confirmed in the Immunochip analysis.
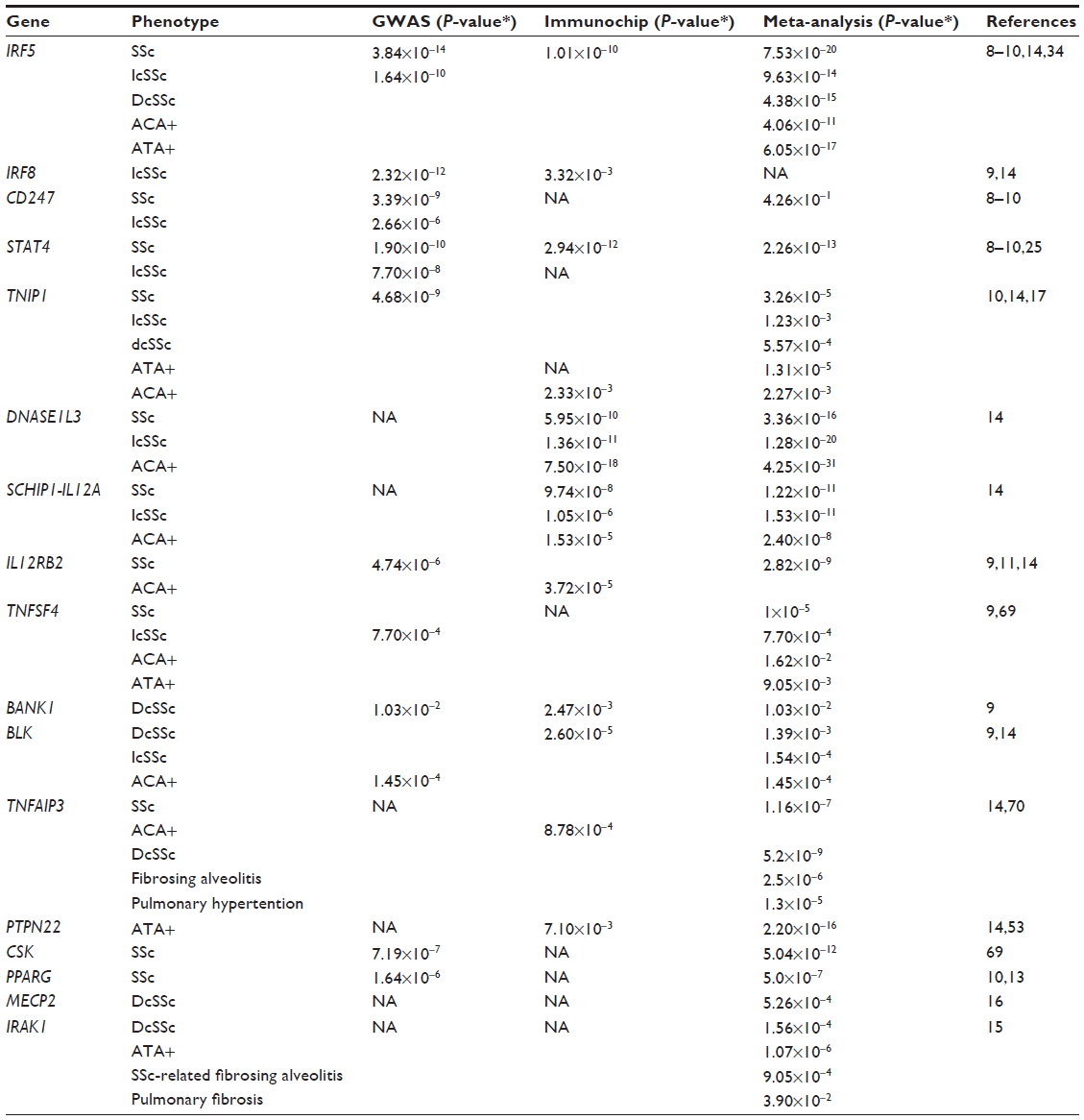 | Table 2 Selected relevant susceptibility alleles in the non-MHC region found with GWAS, Immunochip, and meta-analysis in SSc |
In addition, two genes on the X chromosome have been associated with SSc. These include a polymorphism in the interleukin (IL)-1 receptor-associated kinase 1 gene (IRAK1) associated with SSc-related pulmonary fibrosis, and the methyl-CpG-binding protein 2 gene (MECP2) associated with diffuse SSc.15,16 However, there were discrepant results for PSORS1C1 and RHOB polymorphisms that were not confirmed in an independent cohort.10,17
Together, these studies have clearly established that SSc is a complex autoimmune disease with multiple genetic variants in different loci. Many of these genes are also known to be risk factors for other autoimmune diseases, including rheumatoid arthritis and SLE.18–20 SSc and SLE are two archetypal systemic autoimmune diseases sharing some features, like antinuclear antibodies, but exhibiting very different clinical phenotypes. In order to gain additional insight into the genetic basis of these two diseases, a pan-meta-analysis of two GWASs in SSc and SLE cohorts was performed. The sample size included a total of 21,109 subjects (6,835 cases and 14,274 controls). This analysis determined that the SLE susceptibility loci PXK (PX domain containing serine/threonine kinase) and JAZF1 (juxtaposed with another zinc finger gene 1) shared susceptibility loci with SSc. This study also validated a new KIAA0319-like gene (KIAA0319L) as a susceptibility locus for SSc and SLE. In addition, the gene product of KIAA0319L was found to be over expressed in peripheral blood cells from both SSc and SLE patients, suggesting that this polymorphism was related to immune function.21
These findings confirmed and added new evidence for the strong autoimmune genetic component underlying SSc pathogenesis and further reinforced the concept that there are shared genetic and pathogenic mechanisms among autoimmune diseases.
Type I interferon pathway gene variants associated with SSc and relevant animal studies
It is widely known and accepted that type I interferons (IFNs) are important key regulators of host defense in the innate immune system. IFN regulatory factors are transcriptional regulators of type I IFNs, and IFN-inducible genes and play a pivotal role in regulation of many facets of innate and adaptive immune responses.22 Both SSc GWAS reports and the Immunochip study confirmed genetic variants of IRF genes as susceptibility loci for SSc (Tables 2 and 3). Based on its unique contributory role in type I IFN pathways in the immune system, IRF genes and their protein products may represent valuable targets for therapeutic interventions in autoimmune diseases, including SSc.
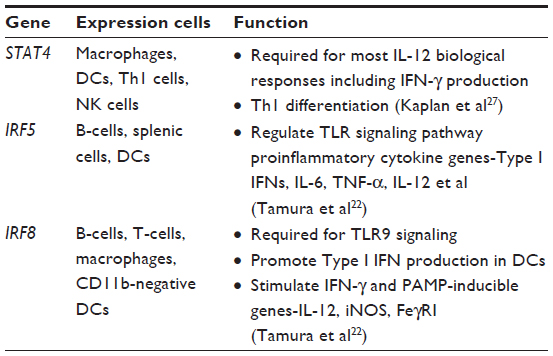 | Table 3 Interferon pathway genes associated with human SSc |
Animal models are very useful to test a specific hypothesis about disease pathogenesis in vivo. Although animal models that exhibit all aspects of SSc are not currently available, several general SSc mouse models are well characterized and widely used, as noted in Table 4. More specific mouse models, developed by forward genetic studies of experimentally induced mutations, can selectively test for the role of genes and proteins that are relevant to the corresponding human disease. Some of these mouse models with specific targeted deletions in type I interferon pathway genes are described in Table 5.
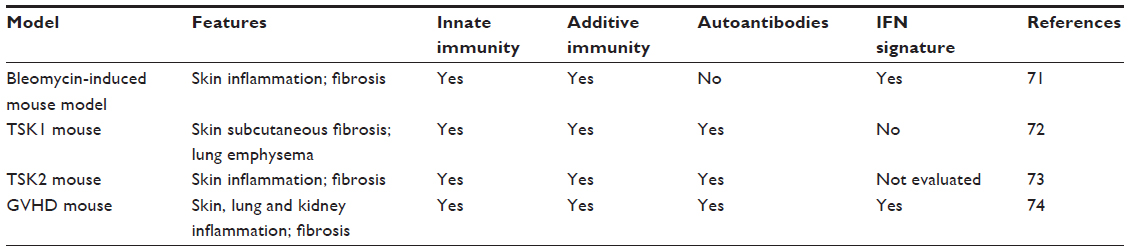 | Table 4 Overview of general mouse models in SSc |
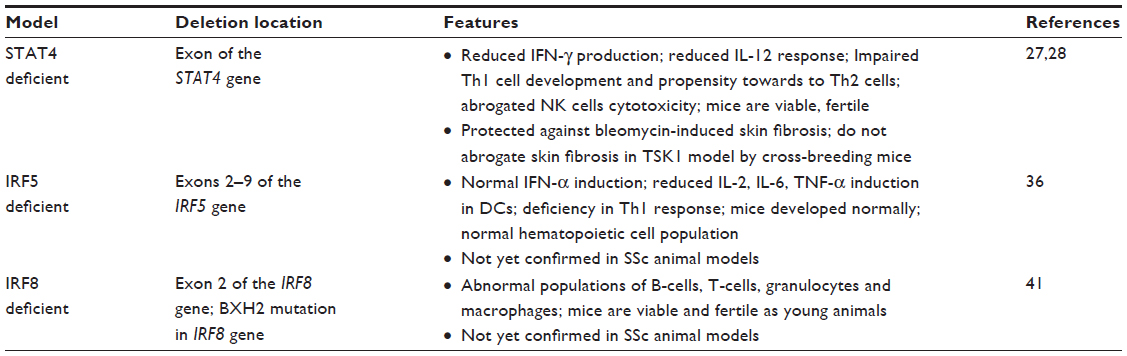 | Table 5 Interferon pathway gene knockout in mouse model |
STAT4
STAT4 is one of the seven members of the signal transducer and activator of transcription (STAT) family, which are important transcription factors for T-cell differentiation and T-cell receptor signaling pathways.23 It is expressed in macrophages, dendritic cells (DCs), T helper (Th)1 cells, and natural killer (NK) cells. STAT4 plays a key role in type I IFN receptor signaling by being activated and trans located to the nucleus after type I IFN receptor ligation.24 It is also required for IFN-γ production and Th1 cell differentiation (see Table 3). Candidate gene studies in European and Japanese cohorts and both large SSc GWASs have identified and validated the association between SSc with variants in the STAT4 gene.8–10,25 In addition, the STAT4 SNP rs7574865 and the IRF5 SNP rs2004640 were observed to have additive effects in terms of susceptibility to both SSc and SSc-related fibrosing alveolitis/interstitial lung disease.26 In an animal study, STAT4-deficient (stat4-/-) mice showed reduced IFN-γ production and reduced IL-12 response in lymphocytes, along with an increased propensity toward the development of Th2 cells.27 The role of STAT4 deficiency has been studied in two murine models of SSc: the bleomycin model and the tight skin 1 (TSK1) model.28 STAT4-deficient (stat4-/-) mice showed protection against the inflammatory and fibrotic processes in the bleomycin-induced SSc animal model. However, when the STAT4-deficient mice were crossed with the non inflammatory TSK1 model to generate TSK1 mice deficient for STAT4 (stat4-/-/tsk1), collagen deposition was not noted to be decreased. These findings suggested that the transcription factor STAT4 exerts its potent profibrotic effects by controlling T-cell activation and proliferation with subsequent inflammatory cytokine release. These findings also underscore the important role of STAT4 implied by the STAT4 genetic association with SSc.28
Further fine-mapping and functional studies are crucial for elucidating the role of genetic variants in the IFN signaling pathway of the immune system in the pathogenesis of SSc.
IRF5
As noted above, the IRF5 gene codes for IFN regulatory factor 5 (IRF5), which is a transcription factor that induces the transcription of genes involved in the immune response, including type 1 IFN, macrophage inflammatory protein-1α, and -β, 2,5-oligoadenylate synthetase, protein kinase R, pre-B-cell colony enhancing factors, and IFN-induced hepatitis C-associated micro tubular aggregate proteins.29,30 IRF5 negatively regulates cell growth and proapoptotic genes, including p21, Bak, DAP kinase 2, and Bax.30 It is mostly expressed in B-cells, splenic cells and DCs.22 Genetic variants of IRF5 were found to be associated with SSc susceptibility in multiple cohorts.8,26,31–34 This may have functional significance, as the minor allele of the IRF5 SNP rs4728142 was shown to be predictive of longer survival and milder pulmonary involvement in two independent SSc cohorts.35 This minor allele was also associated with lower IRF5 transcript expression in monocytes from SSc patients and controls, suggesting functional relevance of SNP rs4728142, or its association with other SNPs, for IRF5 expression. A recent study by Carmona et al34 revealed that IRF5 SNP haplotype blocks containing three haplotypic risk alleles (including rs4728142, rs2004640, rs10488631) showed strong association with overall SSc and was not associated with any specific sub-phenotype.
Animal studies have shown that mutation at exons 2–9 of the IRF5 gene in mice (resulting in irf5-/- null mice) caused increased sensitivity to viral infection and lower expression levels of type I IFN in the serum. IFN production was impaired in the toll-like receptor ligand-stimulated macrophages from these IRF5 mutant mice.36 These studies demonstrate the important role of IRF5 in type I IFN function leading to immune-system abnormalities, which could be relevant to SSc pathogenesis.34 Further studies are needed to explore the functional role of IRF5 in SSc animal models such as the bleomycin model or the TSK1 mouse.
IRF8
The IRF8 gene product is an immune cell-specific IRF family member that is mostly expressed in macrophages, DCs, and B-cells.22 The IRF8 SNP rs11642873 was identified and confirmed as a risk factor for limited cutaneous systemic sclerosis and ACA-positive SSc in a large GWAS and meta-analysis conducted in European and North American cohorts.9 The subsequent Immunochip study identified SNP rs16941172 in the IRF8 gene as being highly associated with the ACA-positive sub phenotype of SSc.14 Another independent study in a Japanese population further added evidence that the IRF8 gene is a susceptibility locus for SSc.37 These studies support involvement of IRF8-expressing innate immune cells including B-cells, DCs, and macrophages in disease development.
Animal studies further support a contributory role for IRF8 in the pathogenesis of autoimmune disease (Table 4). BXH2 mice carry a point mutation in the IRF8/Icsbp gene that changes arginine to cysteine in position 294 (IRF8 R294C).38 The mutation is within the IRF association domain important for the interaction of IRF8 with partner proteins.39,40 The IRF8 R294C mutation abolished the development of CD8 DCs without impairing plasmacytoid DC development, and it eliminated production of some inflammatory cytokines, while retaining that of type I IFNs.41 These studies demonstrated the complex role of IRF8 in DC and monocyte differentiation and immune responses. Further research efforts utilizing SSc animal models are needed to investigate a functional role of IRF8 in human SSc.
Other selected gene variants in human SSc and related animal models
NOTCH4
The NOTCH4 gene codes for the neurogenic locus notch homolog 4 (Notch4) protein, which is a transmembrane receptor that plays a key role in a variety of developmental processes by controlling cell fate decisions. Notch4 is mostly expressed in blood vessels and the notch signaling pathway controls key functions in vascular smooth muscle and endothelial cells, which may be relevant to the microvascular damage in SSc. Although the gene resides in the MHC region of chromosome 6, the NOTCH4 polymorphism is associated with SSc independently from HLA genes or alleles. A recent study has identified a consistent association of the NOTCH4 gene with both ACA-positive and ATA-positive subgroups in four GWAS SSc cohorts (German, Spanish, Dutch, and US) independent of the HLA associations.9 In addition, NOTCH4 polymorphisms have been associated with other autoimmune disorders, including type I diabetes,42 rheumatoid arthritis,43 and SLE.44,45
In an animal study by Villa and colleagues in 2001,46 Notch4 was the only receptor expressed in the mouse endothelium among all of the four Notch family receptors. Furthermore, mouse gain-of-function and loss-of-function studies revealed that NOTCH1 plus NOTCH4 knockouts resulted in embryonic lethality with vascular remodeling defects.47 Although mice deficient in Notch4 alone were viable and fertile,47 angiogenesis was disrupted in these mice, suggesting that Notch signaling plays a more important role in angiogenesis. Endothelial cell-specific expression of an activated form of Notch4 driven by the flk1-promoter led to embryonic lethality with abnormal vessel structure and patterning in mice,48 whose phenotype is similar to that seen in Notch1/Notch4-deficient mice.47 The similar vascular phenotypes observed in both the transgenic (gain-of-function) and the knockout mice (loss-of-function) suggest that there may be an appropriate balance needed in Notch expression levels. To date, how the Notch4 signaling pathway is involved in the pathogenesis of SSc is unclear. Further investigation including NOTCH4 gene manipulation in different SSc mouse models studies may provide better understanding of this pathway.
The search for causal variants
For the most part, the multiple SNPs that have been associated with SSc (and several other autoimmune diseases) are not in coding regions or in known regulatory regions, and therefore it is not clear how these variants affect downstream pathways resulting in disease. However, there are two exceptions to this situation, which are functional variants believed responsible for the PTPN22 and DNASE1L3 associations.
PTPN22
The PTPN22 gene encodes protein tyrosine phosphatase non receptor 22 (PTPN22) which is an intracellular protein tyrosine phosphatase. Polymorphisms in this gene are strongly associated with susceptibility to several autoimmune diseases, including type 1 diabetes, rheumatoid arthritis, and SLE.49–51 The risk allele of PTPN22 results in the exchange of arginine residue at position 620 to tryptophan (R620W). The PTPN22 R620WSNP was also significantly associated with the ATA-positive subset of SSc in large cohort case–control studies.52,53 The Arg620Trp substitution increases the intrinsic lymphoid-specific phosphatase activity of PTPN22, which reduces the threshold for T-cell receptor signaling and thus promotes autoimmunity.54
The corresponding homologue in mice is the PEST (proline–glutamic acid–serine–threonine) domain-enriched tyrosine phosphatase (PEP).55–58 Recently, several animal models of the PEP gene have been extensively investigated. PEP knockout mouse,59 PEP 619W knockin mouse60,61 and PEP knockdown mouse62 studies were established.59,63 In particular, PTPN22 619W mouse mutation is the homologue for the human Arg620Trp substitution and results in spontaneous autoimmunity in susceptible mouse strains, essentially replicating the risk effect of the PTPN22 620W in human autoimmune diseases. Furthermore, findings from PTPN22 genetic mouse models shed new light on both cellular as well as molecular mechanisms of the effect of PTPN22 on adaptive and innate immunity.63
DNASE1L3
The DNASE1L3 gene codes for deoxyribonuclease 1-like 3, a member of the human DNase1 family. It is expressed primarily in liver and spleen and is secreted by macrophages.64 As one of three human homologs of DNase1, DNASE1L3 functions as an endonuclease capable of cleaving both single- and double-stranded DNA. Dnase1 is the major nuclease present in serum, urine, and secretions and is believed to be responsible for the removal of DNA from nuclear antigens at sites of high cell turnover. The non synonymous rs35677470 SNP in exon 8 of this gene results in a substitution of Arg/Cys at the amino acid position 206 of the protein (R206C). An in vitro study indicated that this substitution results in absent DNase activity.65 Recent GWASs and Immunochip analysis identified and confirmed DNASE1L3 genelocus as a risk factor for autoimmune disease, including rheumatoid arthritis, SLE, and SSc.14,66,67 Furthermore, animal studies revealed that DNASE1L3 deficiency in the Murphy Roths Large (MRL) and New Zealand black x New Zealand white (NZB/W) F1 murine models of SLE resulted in increased susceptibility to and severity of SLE.65 DNASE1 knockout mice showed the presence of antinuclear antibodies, deposition of immune complexes in glomeruli and full-blown glomerulonephritis in a Dnase1 dose-dependent manner.68 Altogether, these data suggest a link between defective apoptotic DNA breakdown and the production of autoantibodies and autoimmunity. Further studies to explore regulatory mechanism of DNASE1L3 utilizing animal models in SSc may lead to a better understanding of the pathogenesis, new diagnostic methods, and new therapeutic approaches to SSc and other autoimmune diseases.
Conclusion
To date, only a small proportion of the observed heritability in SSc has been explained. Multiple SSc genetic approaches, including candidate gene studies, the two large GWASs, and the Immunochip studies, have firmly established and confirmed the association of SSc with multiple non-MHCloci including STAT4, IRF5, IRF8, CD247, BANK1, TNFAIP3, TNIP1, IL12RB2, CSK, TNFSF4, BLK, DNASE1L3, SCHIP1-IL12A, and ATG5, as well as HLA-DQA1/B1, HLA-DOA, HLA-DPB1, HLA-DRB1, NOTCH4, TAP2, and MICA/B genes within the MHC region. With few exceptions, such as PTPN22 and DNASE1L3, the causal variants responsible for these disease associations are not known. In addition, the functional relevance of the identified polymorphisms is not clear. Animal studies manipulating the SSc susceptibility gene homologues are needed to identify and characterize the new gene variants and for further confirmatory studies of the “missing heritability” in the larger scale of GWAS and family studies. Interaction between multiple susceptibility alleles of genes and gene-environmental interaction may have greater effects in pathogenesis of SSc and other autoimmune diseases. Animal model studies not only significantly extend our understanding on the mechanism of disease progress, but also may provide new ideas and tools for identifying novel therapeutic target genes and molecules for human diseases.
Acknowledgments
This work was supported by Scleroderma Foundation New Investigator Award (Wu M), National Institutes of Health (NIH), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Centers of Research Translation (CORT) P50AR054144, NIH/NIAMS Scleroderma Family Registry and DNA Repository grant N01-AR02251, NIH National Center for Clinical and Translational Sciences grant 3UL1RR024148, and DOD Congressionally Directed Medical Research Program W81XWH-13-1-0452 (Mayes MD).
Disclosure
The authors report no conflicts of interest in this work.
References
Sapadin AN, Esser AC, Fleischmajer R. Immunopathogenesis of scleroderma – evolving concepts. Mt Sinai J Med. 2001;68(4–5):233–242. | |
Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol. 2012;24(2):165–170. | |
Arnett FC, Cho M, Chatterjee S, Aguilar MB, Reveille JD, Mayes MD. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001;44(6):1359–1362. | |
Zhou X, Tan FK, Xiong M, Arnett FC, Feghali-Bostwick CA. Monozygotic twins clinically discordant for scleroderma show concordance for fibroblast gene expression profiles. Arthritis Rheum. 2005;52(10):3305–3314. | |
Frech T, Khanna D, Markewitz B, Mineau G, Pimentel R, Sawitzke A. Heritability of vasculopathy, autoimmune disease, and fibrosis in systemic sclerosis: a population-based study. Arthritis Rheum. 2010;62(7):2109–2116. | |
Martín JE, Bossini-Castillo L, Martín J. Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012;131(7):1023–1037. | |
Mayes MD. The genetics of scleroderma: looking into the postgenomic era. Curr Opin Rheumatol. 2012;24(6):677–684. | |
Radstake TR, Gorlova O, Rueda B, et al; Spanish Scleroderma Group. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42(5):426–429. | |
Gorlova O, Martin JE, Rueda B, et al; Spanish Scleroderma Group. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 2011;7(7):e1002178. | |
Allanore Y, Saad M, Dieudé P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7(7):e1002091. | |
Bossini-Castillo L, Martin JE, Broen J, et al; Spanish Scleroderma Group. A GWAS follow-up study reveals the association of the IL12RB2 gene with systemic sclerosis in Caucasian populations. Hum Mol Genet. 2012;21(4):926–933. | |
Martin JE, Broen JC, Carmona FD, et al; Spanish Scleroderma Group. Identification of CSK as a systemic sclerosis genetic risk factor through Genome Wide Association Study follow-up. Hum Mol Genet. 2012;21(12):2825–2835. | |
López-Isac E, Bossini-Castillo L, Simeon CP, et al; Spanish Scleroderma Group. A genome-wide association study follow-up suggests a possible role for PPARG in systemic sclerosis susceptibility. Arthritis Res Ther. 2014;16(1):R6. | |
Mayes MD, Bossini-Castillo L, Gorlova O, et al; Spanish Scleroderma Group. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet. 2014;94(1):47–61. | |
Dieudé P, Bouaziz M, Guedj M, et al. Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum. 2011;63(12):3979–3987. | |
Carmona FD, Cénit MC, Diaz-Gallo LM, et al; Spanish Scleroderma Group. New insight on the Xq28 association with systemic sclerosis. Ann Rheum Dis. 2013;72(12):2032–2038. | |
Bossini-Castillo L, Martin JE, Broen J, et al; Spanish Scleroderma Group. Confirmation of TNIP1 but not RHOB and PSORS1C1 as systemic sclerosis risk factors in a large independent replication study. Ann Rheum Dis. 2013;72(4):602–607. | |
Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104(16):6758–6763. | |
Graham RR, Kozyrev SV, Baechler EC, et al; Argentine and Spanish Collaborative Groups. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38(5):550–555. | |
Sigurdsson S, Nordmark G, Göring HH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76(3):528–537. | |
Martin JE, Assassi S, Diaz-Gallo LM, et al; Spanish Scleroderma Group; SLEGEN consortium; US Scleroderma GWAS group; BIOLUPUS. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet. 2013;22(19):4021–4029. | |
Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. | |
Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2(11):536–550. | |
Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res. 2005;31(3):231–242. | |
Rueda B, Broen J, Simeon C, et al. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet. 2009;18(11):2071–2077. | |
Dieudé P, Guedj M, Wipff J, et al. STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum. 2009;60(8):2472–2479. | |
Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382(6587):174–177. | |
Avouac J, Fürnrohr BG, Tomcik M, et al. Inactivation of the transcription factor STAT-4 prevents inflammation-driven fibrosis in animal models of systemic sclerosis. Arthritis Rheum. 2011;63(3):800–809. | |
Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. 2004;279(43):45194–45207. | |
Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM. Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res. 2003;63(19):6424–6431. | |
Dieudé P, Wipff J, Guedj M, et al. BANK1 is a genetic risk factor for diffuse cutaneous systemic sclerosis and has additive effects with IRF5 and STAT4. Arthritis Rheum. 2009;60(11):3447–3454. | |
Dieudé P, Guedj M, Wipff J, et al. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: a new perspective for pulmonary fibrosis. Arthritis Rheum. 2009;60(1):225–233. | |
Ito I, Kawaguchi Y, Kawasaki A, et al. Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum. 2009;60(6):1845–1850. | |
Carmona FD, Martin JE, Beretta L, et al; Spanish Scleroderma Group. The systemic lupus erythematosus IRF5 risk haplotype is associated with systemic sclerosis. PLoS One. 2013;8(1):e54419. | |
Sharif R, Mayes MD, Tan FK, et al. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis. 2012;71(7):1197–1202. | |
Takaoka A, Yanai H, Kondo S, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434(7030):243–249. | |
Terao C, Ohmura K, Kawaguchi Y, et al. PLD4 as a novel susceptibility gene for systemic sclerosis in a Japanese population. Arthritis Rheum. 2013;65(2):472–480. | |
Turcotte K, Gauthier S, Tuite A, Mullick A, Malo D, Gros P. A mutation in the Icsbp1 gene causes susceptibility to infection and a chronic myeloid leukemia-like syndrome in BXH-2 mice. J Exp Med. 2005;201(6):881–890. | |
Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem. 2007;282(28):20065–20069. | |
Levi BZ, Hashmueli S, Gleit-Kielmanowicz M, Azriel A, Meraro D. ICSBP/IRF-8 transactivation: a tale of protein-protein interaction. J Interferon Cytokine Res. 2002;22(1):153–160. | |
Tailor P, Tamura T, Morse HC, Ozato K. The BXH2 mutation in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood. 2008;111(4):1942–1945. | |
Valdes AM, Thomson G; Type 1 Diabetes Genetics Consortium. Several loci in the HLA class III region are associated with T1D risk after adjusting for DRB1-DQB1. Diabetes Obes Metab. 2009;11 Suppl 1:46–52. | |
AlFadhli S, Nanda A. Genetic evidence for the involvement of NOTCH4 in rheumatoid arthritis and alopecia areata. Immunol Lett. 2013; 150(1–2):130–133. | |
Rioux JD, Goyette P, Vyse TJ, et al; International MHC and Autoimmunity Genetics Network. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci U S A. 2009;106(44):18680–18685. | |
Morris DL, Taylor KE, Fernando MM, et al; International MHC and Autoimmunity Genetics Network; Systemic Lupus Erythematosus Genetics Consortium. Unraveling multiple MHC gene associations with systemic lupus erythematosus: model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am J Hum Genet. 2012;91(5):778–793. | |
Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108(1–2):161–164. | |
Krebs LT, Xue Y, Norton CR, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14(11):1343–1352. | |
Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci U S A. 2001;98(10):5643–5648. | |
Begovich AB, Carlton VE, Honigberg LA, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75(2):330–337. | |
Kyogoku C, Langefeld CD, Ortmann WA, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet. 2004;75(3):504–507. | |
Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36(4):337–338. | |
Gourh P, Tan FK, Assassi S, et al. Association of the PTPN22 R620W polymorphism with anti-topoisomerase I- and anticentromere antibody-positive systemic sclerosis. Arthritis Rheum. 2006;54(12):3945–3953. | |
Dieudé P, Guedj M, Wipff J, et al. The PTPN22 620W allele confers susceptibility to systemic sclerosis: findings of a large case-control study of European Caucasians and a meta-analysis. Arthritis Rheum. 2008;58(7):2183–2188. | |
Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006;18(4):207–213. | |
Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood. 1999;93(6):2013–2024. | |
Matthews RJ, Bowne DB, Flores E, Thomas ML. Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol Cell Biol. 1992;12(5):2396–2405. | |
Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189(1):111–121. | |
Gjörloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. 1999;29(12):3845–3854. | |
Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal. 2012;5(252):ra87. | |
Dai X, James RG, Habib T, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013;123(5):2024–2036. | |
Zhang J, Zahir N, Jiang Q, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43(9):902–907. | |
Zheng P, Kissler S. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes. 2013;62(3):896–904. | |
Zheng J, Petersen F, Yu X. The role of PTPN22 in autoimmunity: learning from mice. Autoimmun Rev. 2014;13(3):266–271. | |
Baron WF, Pan CQ, Spencer SA, Ryan AM, Lazarus RA, Baker KP. Cloning and characterization of an actin-resistant DNase I-like endonuclease secreted by macrophages. Gene. 1998;215(2):291–301. | |
Ueki M, Takeshita H, Fujihara J, et al. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin Chim Acta. 2009;407(1–2):20–24. | |
Al-Mayouf SM, Sunker A, Abdwani R, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43(12):1186–1188. | |
Eyre S, Bowes J, Diogo D, et al; Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate; Wellcome Trust Case Control Consortium. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44(12):1336–1340. | |
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. | |
Martin JE, Broen JC, Carmona FD, et al; Spanish Scleroderma Group. Identification of CSK as a systemic sclerosis genetic risk factor through Genome Wide Association Study follow-up. Hum Mol Genet. 2012;21(12):2825–2835. | |
Dieudé P, Guedj M, Wipff J, et al. Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis. 2010;69(11):1958–1964. | |
Yamamoto T, Takagawa S, Katayama I, et al. Animal model of sclerotic skin. I: Local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol. 1999;112(4):456–462. | |
Manne J, Markova M, Siracusa LD, Jimenez SA. Collagen content in skin and internal organs of the tight skin mouse: an animal model of scleroderma. Biochem Res Int. 2013;2013:436053. | |
Christner PJ, Peters J, Hawkins D, Siracusa LD, Jiménez SA. The tight skin 2 mouse. An animal model of scleroderma displaying cutaneous fibrosis and mononuclear cell infiltration. Arthritis Rheum. 1995;38(12):1791–1798. | |
Ruzek MC, Jha S, Ledbetter S, Richards SM, Garman RD. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 2004;50(4):1319–1331. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.