")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
In vitro transfection of bone marrow-derived dendritic cells with TATp-liposomes
Authors Pappalardo JS, Langellotti CA, Di Giacomo S, Olivera V, Quattrocchi V, Zamorano PI, Hartner WC, Levchenko TS, Torchilin VP
Received 23 August 2013
Accepted for publication 10 November 2013
Published 13 February 2014 Volume 2014:9(1) Pages 963—973
DOI https://doi.org/10.2147/IJN.S53432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Juan Sebastián Pappalardo,1–3 Cecilia A Langellotti,2 Sebastián Di Giacomo,1 Valeria Olivera,1 Valeria Quattrocchi,2 Patricia I Zamorano,1,2 William C Hartner,3 Tatyana S Levchenko,3 Vladimir P Torchilin3
1Virology Institute, Center for Research in Veterinary and Agronomic Sciences, National Institute for Agricultural Technology (INTA), Hurlingham, BA, Argentina; 2National Council for Scientific and Technical Research (CONICET), Autonomous City of Buenos Aires, Argentina; 3Center for Pharmaceutical Biotechnology and Nanomedicine, Northeastern University, Boston, MA, USA
Abstract: Dendritic cells (DC) are antigen-presenting cells uniquely capable of priming naïve T cells and cross-presenting antigens, and they determine the type of immune response elicited against an antigen. TAT peptide (TATp), is an amphipathic, arginine-rich, cationic peptide that promotes penetration and translocation of various molecules and nanoparticles into cells. TATp-liposomes (TATp-L) used for DC transfection were prepared using TATp derivatized with a lipid-terminated polymer capable of anchoring in the liposomal membrane. Here, we show that the addition of TATp to DNA-loaded liposomes increased the uptake of DNA in DC. DNA-loaded TATp-L increased the in vitro transfection efficiency in DC cultures as evidenced by a higher expression of the enhanced green fluorescent protein and bovine herpes virus type 1 glycoprotein D (gD). The de novo synthesized gD protein was immunologically stimulating when transfections were performed with TATp-L, as indicated by the secretion of interleukin 6.
Keywords: dendritic cell transfection, green fluorescent protein, bovine herpes virus 1 glycoprotein D, liposomes, TAT peptide, interleukin 6
Introduction
Dendritic cells (DC) are professional antigen-presenting cells (APC), central to the immune response. DC initiate primary immune responses in vivo and are key linkers between innate and adaptive immunity. These cells function in the presentation of antigens, a critical role that leads either to an immune response or anergy toward the antigen. DC are capable of priming naïve T cells and cross-presenting antigens and determining the type of immune response.1,2
Peptides, derived from endogenous antigens degraded in the cytosolic space via the proteasome system, are presented mainly by major histocompatibility complex (MHC) class I molecules and initiate cellular immunity through cytotoxic T lymphocyte (CTL) activation, whereas peptides from extracellularly endocytosed antigens, which are degraded in the lysosomes, are presented mainly by MHC class II molecules followed by activation of antibody (Ab)-secreting B cells.3
One of the challenges in vaccine development is the activation of CTL-mediated responses. However, it is difficult to elicit a CTL-mediated response when using inactivated antigens or peptides since they are captured and processed by APC as exogenous antigens.4 Still, a CTL-mediated response may become possible following a DNA transfection that results in the de novo synthesis of the desired protein, which is then presented as an endogenous antigen. In addition, Ab-mediated protection against cancer and some viral infections is not as efficient as cellular immunity. In herpes virus infections, both the eradication of infected cells and the inactivation of viral particles are crucial to successful therapies.5–14
The genetic manipulation of DC should prove very useful for vaccination or cancer therapy.15–17 Good levels of transfection of DC in humans and mice have been achieved in vitro with different techniques. The electroporation method is tricky and is used preferably with mature DC.18,19 Synthetic transfection reagents are inefficient for gene transfer to DC and are usually too toxic for use in vivo.20 Viral vectors themselves, even when efficient, also present the risk that viral products will interfere with normal cell functions.21
Liposomes are lipid bilayered nanovesicles, which can serve as carriers for DNA, proteins, and other antigens, as well as drugs. They are now widely used for cancer research and treatment, and in experimental gene therapy. Liposomes modified with cell-penetrating peptides, such as TAT peptide (TATp-L), can be prepared by attachment of TATp to polymer-lipid conjugates, such as polyethylene glycol-phosphatidylethanolamine (PEG-PE), followed by incorporation of the TATp-PEG-PE into the liposome membrane with this lipid moiety. Cell-penetrating peptides represent amphipathic, arginine-rich, cationic peptides that penetrate and translocate into the cell.22,23
TATp-L would seem to be good candidates for DC transfection. TATp-L characterization, its properties, and the ability to improve transfection in various cell lines and spleen-derived macrophage primary cultures have been demonstrated in our previous studies.24–27 The activation of DC has been shown to be especially important in the generation of an immune response and is currently considered a requirement/necessary for the generation of a good CTL response for the prevention of certain infectious diseases or in the treatment of cancer patients.
Here, we show that the modification of liposomes with TATp increases the uptake and internalization of liposomes in DC. In addition, TATp-L increase the in vitro transfection efficiency by DC cultures as evidenced by an enhanced green fluorescent protein (EGFP) and bovine herpes virus type 1 (BoHV-1) glycoprotein D (gD) expression. Moreover, the de novo synthesized gD is immunologically active when transfections are performed with TATp-L, as indicated by the secretion of interleukin (IL)-6.
Materials and methods
Materials
Egg phosphatidylcholine, cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG2K-DSPE), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh-DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Polyoxyethylene (MW 3400)-bis(para-nitrophenyl carbonate) (PEG3.4K-(pNP)2) was purchased from SunBio (Anyang City, South Korea). CL-4B sepharose, triethylamine, tryptone, yeast extract, and components of buffer solutions were from Sigma-Aldrich (St Louis, MO, USA). Lipofectin®, Lipofectamine®, and Hoechst 33342 were from Invitrogen Life Sciences (Life Technologies, Carlsbad, CA, USA). TATp (11-mer: YGRKKRRQRRR, 1.56 KDa) was prepared by Invitrogen Life Sciences. Roswell Park Memorial Institute (RPMI)-1640 medium and heat-inactivated fetal bovine serum (FBS) were supplied by Cellgro (Mediatech Inc, Manassas, VA, USA). Fluorescence-free glycerol-based mounting media (Fluoromount-G™) was from Southern Biotechnology Associates (Birmingham, AL, USA). The pEGFP-N1 plasmid designed for eukaryotic cell expression of the EGFP was obtained from Elim Biopharmaceuticals Inc (Hayward, CA, USA). The eukaryotic cell expression systems with cytomegalovirus promoter pCI-neo (pCIneo), and BamHI and EcoRI restriction enzymes were from Promega (Madison, WI, USA). The pCIgDA plasmid for the eukaryotic cell expression of the BoHV-1 (GenBank Accession number Emb AJ004801) gD was designed at the Instituto Nacional de Tecnología Agropecuaria (INTA) (Hurlingham, Argentina).28 Competent Escherichia coli DH5α were from INTA. A transfection grade purification Plasmid Mega Kit was purchased from Qiagen (Valencia, CA, USA). Mouse monoclonal Ab (mAb) against gD was kindly provided by Dr Lorne Babiuk29,30 (Vaccine and Infectious Disease Organization [VIDO], University of Saskatchewan, Saskatoon, SK, Canada). Molecular weight marker kaleidoscope prestained standards were from Bio-Rad Laboratories (Hercules, CA, USA). Female 6–8 week old BALB/c mice were purchased from Charles River Laboratories (Wilmington, MA, USA). Lab-Tek 4-, 8-, and 16-well cell culture chambers were from Nunc-Thermo Fisher Scientific (Rochester, NY, USA). Murine recombinant granulocyte-macrophage colony-stimulating factor (mr-GM-CSF) was obtained from R&D Systems (Minneapolis, MN, USA). Luria Bertani medium was prepared according to Qiagen Plasmid Mega Kit datasheet specifications. Fluorescein isothiocyanate (FITC)-labeled anti-mouse CD11c (cluster of differentiation 11c), peridinin chlorophyll cyanine 5.5 (PerCP-Cy5.5)-labeled anti-mouse CD11c, phycoerythrin (PE)-labeled anti-IAd (MHC class II), PE-labeled anti-mouse Gr-1 (granulocyte-differentiation antigen-1) mAbs, FITC, or PerCP-Cy5.5-labeled Armenian hamster immunoglobulin G (IgG) and PE-labeled rat IgG2b isotype controls were purchased from eBioscience (San Diego, CA, USA). Complete RPMI (comp-RPMI) was prepared with RPMI-1640, 10% FBS, and 5.5×10−5 M 2-mercaptoethanol from Sigma-Aldrich, and penicillin-streptomycin and amphotericin-B mix from Invitrogen Life Sciences. Dendritic cell RPMI (DC-RPMI) was prepared with comp-RPMI plus 20 ng/mL of mr-GM-CSF.
Generation of bone marrow-derived DC
Bone marrow-derived DC were obtained as described in Inaba et al31 with modifications. Briefly, mice were anesthetized, sacrificed by cervical dislocation following a protocol approved by Northeastern University Institutional Animal Care and Use Committee in accordance with Principles of Laboratory Animal Care (NIH publication No 85-23, revised in 1996), and soaked for 10 minutes in 70% ethanol. The femurs and tibias were removed and placed in 70% ethanol for 1 minute. The epiphyses were discarded and the diaphyses were placed in a 5 mL Petri dish with sterile comp-RPMI. Cells were removed by perfusion with media from a syringe with a needle inserted in the widest end of the bone. Collected cells were dispersed with a syringe and centrifuged at 1,200 rpm (315 RCF) for 5 minutes at room temperature (RT) in a Sorvall® RT6000B (Du Pont Company, Wilmington, DE, USA) with H1000B rotor. The cell pellet was resuspended in DC-RPMI at a concentration of 1×106 cells/mL.
The cultures were fed at 48-hour intervals for 6 days by repeating the same procedure. The plates were gently swirled to remove nonadherent granulocytes without dislodging clusters of developing DC that were loosely attached to firmly adherent macrophages. Then 80% of the media was aspirated and removed. Fresh DC-RPMI was added very slowly to avoid disrupting the clusters. At day 8, the cell aggregates were harvested by trypsinization for 5 minutes, followed by centrifugation as above and overnight subculture at 3×105 cells/mL in comp-RPMI. The subcultures were fed at 48-hour intervals from days 10 to 14 with comp-RPMI.
Between days 4 and 14 of culture, cells were tested for specific markers. When more than 60% of the loosely adherent cells expressed the CD11c marker, they were considered ready to use.
Synthesis of pNP-PEG-PE
pNP-PEG-PE was synthesized according to a previously published procedure.32 Approximately 30 μmol of DOPE was dissolved in chloroform to obtain a 50 mg/mL solution and supplemented with 80 μL (circa 2-fold molar excess over PEG3.4K-[pNP]2) of triethylamine. Then 300 μmol of PEG3.4K-(pNP)2 dissolved in 5 mL of chloroform was added to the mixture and the sample was incubated overnight at RT with stirring under argon. The organic solvents were removed using a rotary evaporator. The pNP-PEG3.4K-DOPE micelles were formed in 0.01 M HCl, 0.15 M NaCl using water bath sonication. The micelles were separated from the unbound PEG and the released pNP on a CL-4B column using 0.01 M HCl, 0.15 M NaCl as an eluent. Pooled fractions containing pNP-PEG3.4K-DOPE were freeze-dried, and the pNP-PEG3.4K-DOPE was extracted with chloroform. The latter procedure was repeated twice to ensure the complete removal of NaCl from the preparation. pNP-PEG3.4K-DOPE was stored as a chloroform solution at −80°C.
Synthesis of TATp-PEG-PE
The attachment of TATp to pNP-PEG3.4K-DOPE was carried out according to a previously published procedure.32 Briefly, chloroform was removed from the pNP-PEG3.4K-DOPE solution by rotary evaporation. The pNP-PEG3.4K-DOPE micelles were formed in an Na-citrate buffer, pH 5.1, and mixed with TATp in a borate buffer, pH 8.5. The TATp-PEG-PE sample was incubated overnight at RT, and dialyzed against water to eliminate the unbound TATp and free pNP, freeze-dried and stored at −80°C.
Preparation of cationic liposomes
TATp-L: A lipid film was prepared by rotary evaporation from a mixture of phosphatidylcholine, cholesterol, DOTAP, and TATp-PEG3.4K-DOPE (60:30:10:2 molar ratio) with a 0.25 molar fraction of Rh-DOPE (when visualization was needed) in chloroform. This film was rehydrated for 30 minutes in sterile HEPES-buffered saline (HBS), pH 7.4, at a concentration of 1.78 μg lipid/μL. The suspension was vortexed for 5 minutes, and extruded through a polycarbonate filter (200 nm pore size) with an Avanti Polar Lipids MiniExtruder.
Plain liposomes (plain-L): These liposomes were prepared using PEG2K-DSPE instead of TATp-PEG3.4K-DOPE following the procedure just described.
Liposome sizes were measured by the dynamic light scattering on a Coulter N4 Plus (Coulter Corporation, Miami, FL, USA) submicron particle analyzer. Z-potential was measured with a Brookhaven Instruments Corporation (Holtsville, NY, USA) analyzer.
Preparation of liposome–plasmid complexes
Liposomes were incubated with pEGFP-N1 or pCIgDA overnight at 4°C at the cationic-lipid/DNA molar ratio of 5.7:1. The liposome–plasmid complexes were prepared by incubating 12 μg DNA with 800 μL of liposome suspension (a 0.015 μg DNA/μL liposome suspension). To assess the complex’s stability, a sample of the liposome–plasmid complex was treated with Triton X-100 for 60 minutes at 37°C to release the plasmid from the complex (duplicate sample was treated in phosphate-buffered saline [PBS] at the same conditions as the control) and then subjected to agarose gel electrophoresis. Lipofectin and Lipofectamine with plasmid complexes were prepared according to the manufacturer’s instructions with the same quantities of DNA.
Agarose gel electrophoresis
Electrophoresis was performed with the Invitrogen E-Gel electrophoresis system. A precast 0.8% agarose E-Gel cartridge was pre-run for 2 minutes at 60 V and 500 mA followed by loading of liposomes pretreated with Triton X-100 or PBS (control). The gel was run again for 30 minutes at 60 V and 500 mA and photographed over an ultraviolet box (Photodyne; New Berlin, WI, USA).
Uptake of liposomes by DC
Liposome uptake by DC, their intracellular localization and intracellular fate were studied in DC treated with Rh-liposomes. DC were grown in Lab-Tek chambers at 2×105 DC per well in 8-well chambers, or at 4×105 DC per well in 4-well chambers (equal in surface area to the 24-well plates, used to perform the transfections), and treated with different formulations in serum-free RPMI at 37°C under 5% CO2. The medium was removed, and the cells were washed with sterile PBS, pH 7.4, incubated for 5 minutes, and fresh comp-RPMI immediately added. After 15, 30, 60, and 120 minutes incubation, cell nuclei were stained with Hoechst 33342 for 10 minutes, washed with sterile PBS and resuspended in comp-RPMI after detachment from the chamber. To avoid cell crushing, coverslips with small separators were used to create a thin (0.1 mm) chamber for the cells. No mounting media was used to avoid artifacts. Samples were viewed by epifluorescence microscopy at different planes when necessary with an Eclipse E400 (Nikon Corporation, Tokyo, Japan).
Transfection in vitro
DC were transfected using various formulations of liposome–plasmid complexes. DC were grown in Lab-Tek chambers with 2×105 DC per well in 8-well chambers, or at 4×105 DC per well in 4-well chambers and incubated with different formulations (in the quantity required to deliver 0.6 μg of DNA per 2×105 DC at a DNA concentration of 0.015 μg/μL of added liposomal suspension) for 5 hours in a serum-free RPMI at 37°C under 5% CO2. The same quantity of Lipofectin–pEGFP-N1 or Lipofectamine–pCIgDA complex with the same lipid-to-DNA ratio was used as a positive transfection reagent control. Negative nanovehicle controls were also used (various plasmids plus HBS). After transfection, the medium was removed, and fresh comp-RPMI was added immediately. After 48 hours of reincubation, the cell nuclei were stained with Hoechst 33342 for 10 minutes, washed with sterile PBS, mounted with fluorescence-free glycerol-based mounting medium and viewed by epifluorescence microscopy.
Duplicate samples in 24-well plates were chilled at 4°C for 15 minutes and then trypsinized at 37°C for 10 minutes with constant rocking in order to assure complete detachment of the DC culture that in some cases became strongly attached to the plates after treatment. Cell samples were labeled with PerCp-Cy5.5 anti-mouse DC11c mAb for flow cytometry, without the use of Hoechst 33342 to avoid fluorescence interference.
Flow cytometry
Cells were labeled using fluorochrome-labeled mAbs (eBioscience) to evaluate the expression of cell-surface molecules as previously described.27 Briefly, single-cell suspensions (5×105 cells) were incubated with the indicated (see Materials) antibodies diluted in PBS supplemented with 10% FBS at a concentration of 0.2–0.5 μg per 1×106 cells at 4°C for 30 minutes and washed twice with PBS.
Fluorescence-activated cell sorting analysis was performed using a FACScan™ flow cytometer and CellQuest software (Becton Dickinson, San Jose, CA, USA).
Sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting
Electrophoresis was performed with 10% polyacrylamide sodium dodecyl sulfate (SDS) gels, for 60 minutes at 20 V and 100 mA (PowerPac HC; Bio-Rad Laboratories). Gels were loaded with 20 μL of transfected cell lysate in the loading dye. After running, the gels were transferred to a methylcellulose membrane for 45 minutes at 20 V and 400 mA on a Bio-Rad blotter and labeled with an anti-gD mAb or an anti-BoHV-1 polyclonal Ab. Briefly, the membranes were blocked with PBS with Tween-20 (PBST) plus 3% low fat dehydrated milk (blocking buffer), overnight at 4°C with constant rocking. Membranes were incubated with mouse anti-gD mAb (VIDO) in blocking buffer at an appropriate dilution, for 60 minutes at RT with constant rocking. Membranes were washed three times with PBST and incubated in the same conditions with biotinylated goat anti-mouse IgG (H+L), human serum-adsorbed polyclonal Ab (Kirkegaard and Perry Laboratories, Inc, Gaithersburg, MD, USA). Membranes were washed again in the same way and incubated as previously described with streptavidin-alkaline phosphatase. Membranes were washed five times with PBST and developed with nitro-blue-tetrazolium-5-bromo-4-chloro-3-indolyl-phosphate (NBT-BCIP) (Promega). ImageJ freeware image processing developed at the National Institutes of Health was used for image analysis (http://rsbweb.nih.gov/ij/).
Cytokine enzyme-linked immunosorbent assay
Cytokine concentrations were determined in the supernatants of gDA-transfected DC 48 hours post-transfection. Cytokine determinations were performed by a sandwich enzyme-linked immunosorbent assay (ELISA). Briefly, ELISA plates (Maxisorp; Nunc) were coated with rat-anti-mouse IL-6, IL-10, IL-12, or tumor necrosis factor alpha (TNFα) antibodies (Pharmingen, San Diego, CA, USA) diluted in carbonate-bicarbonate buffer (0.05 M, pH 9.6) and incubated overnight at 4°C. The plates were washed three times with PBST, and blocked with PBS supplemented with 10% FBS for 1 hour at RT. Culture supernatant and standards were added to the plates in duplicate and incubated for 2 hours at 4°C. After three washings, the corresponding biotinylated anti-cytokine Ab was added and incubated for 1 hour at 37°C. The plates were washed, incubated with horseradish-peroxidase-conjugated streptavidin for 1 hour. After re-washing, 3,3′,5,5′-tetramethylbenzidine substrate was added. The absorbance at 450 nm was measured in a Multiskan EX spectrophotometer (LabSystems, Helsinki, Finland). Cytokine concentrations were calculated based on the optical densities obtained with standards.
Statistical analyses
All series of experiments were performed at least three times with the exception of the Western blot (WB), which was performed once.
Student’s t-test was used to compare two groups (plain-L and TATp-L) when considering EGFP transfection efficiency. A one-way ANOVA and a Bonferroni post hoc test were used to compare various treatment effects on IL-6 secretion. A P<0.05 for comparisons was considered significant. Log10 transformation was done when necessary.
Results
Cell culture characterization
Flow cytometry of the bone marrow-derived DC cultures demonstrated that more than 60% of the loosely adherent cells exhibited specific molecular markers for DC (CD11c), which provided a sufficient level of cell purity for these studies (Figure 1). MHC class II marker was present (20%) and Gr-1-positive cells constituted less than 10% of all cells (data not shown).
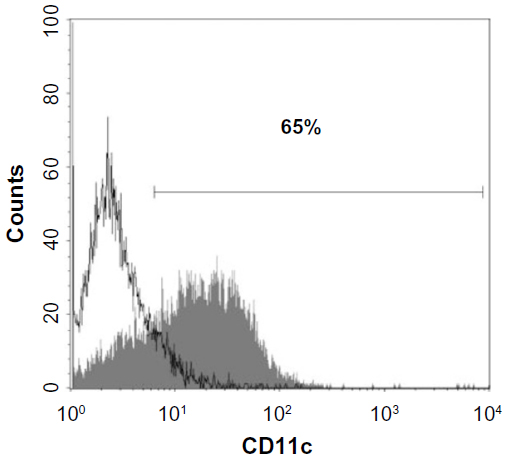 | Figure 1 Evaluation of GM-CSF-stimulated bone marrow-derived cells cultured (loosely attached cells) for the expression of CD11c. |
Liposome characterization
Size measurements showed that more than 90% of liposomes were within the range of 100–250 nm, with the average size of 150–200 nm. ζ-potential values were 4.5 mV for plain-L and 6.9 mV for TATp-L (Table 1). In addition, liposomes were shown to form a stable complex with plasmids that were disaggregated when incubated with detergent, allowing the release of the plasmids (Figure 2).
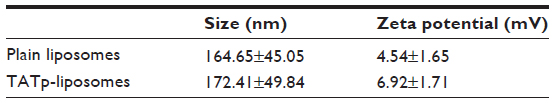 | Table 1 Size and ζ-potential of plain and TATp-liposomes |
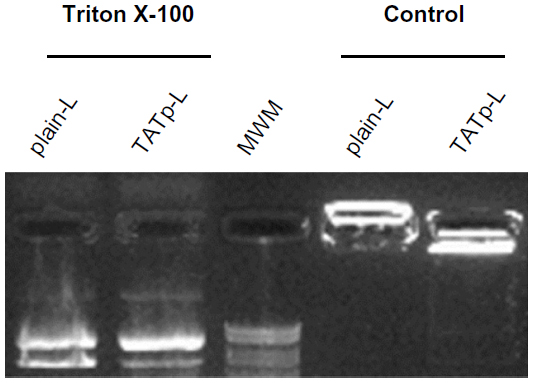 | Figure 2 Evaluation of liposome stability and plasmid release. |
Liposome uptake by DC
This assay allowed the visualization of liposome uptake by cells with liposomes labeled with trace amounts of rhodamine. The liposomes were incubated with DC for 10 minutes. Cells treated with TATp-L showed a strong adherence to liposomes (Figure 3). Furthermore, the penetration of TATp-L into DC was evident from the accumulation of the liposomes in small, well-defined vesicle-like compartments within these cells. Plain-L-treated DC showed faint background surface staining.
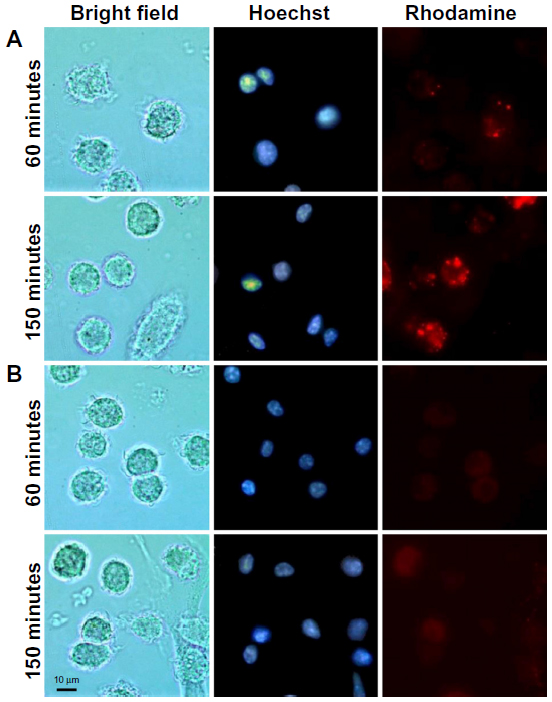 | Figure 3 Fluorescence microscopy of rhodamine-labeled liposomes in DC. |
TATp-liposome penetration of DC
To demonstrate that TATp-L penetrated DC, serial pictures of the treated cells that showed rhodamine fluorescence were taken in two parallel planes (tangential and longitudinal) after 150 minutes of incubation in complete medium. The images clearly demonstrated vesicles with Rh-labeled TATp-L inside cells (Figure 4), which confirmed the enhanced penetration of DC by TATp-L.
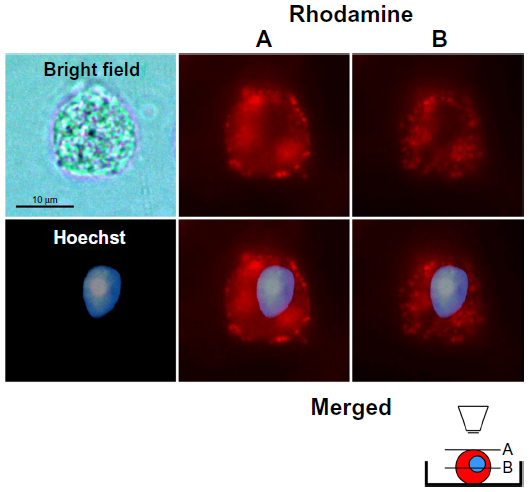 | Figure 4 Dendritic cells after 10-minutes incubation with TATp-L and 150 minutes in normal medium. |
In vitro transfection of DC with TATp-liposomes
After 48 hours of incubation, DC cultures treated with either plain-L or TATp-L loaded with the DNA encoding for the EGFP clearly demonstrated an increased fluorescence in samples with TATp-L (Figure 5). The quantitation of the transfection with cell counts showed a transfection increase of 36% with a TATp-L carrier compared to a plain-L carrier (67%±5.0% versus 31%±6.9%, respectively) (Figure 6A). When replicate samples were analyzed by flow cyto-metry, similar results were obtained. However, when double analysis with specific DC marker versus EGFP expression was performed it was shown that 47% (TATp-L) and 22% (plain-L) were indeed transfected DC (Figure 6B).
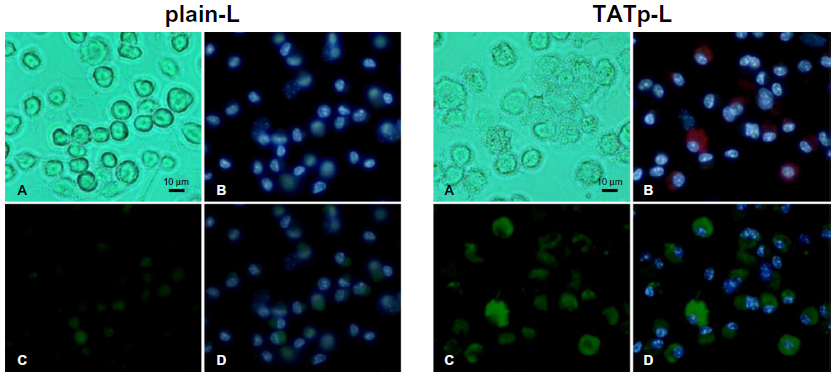 | Figure 5 Fluorescence microscopy of bone-marrow-derived DC 48 hours post-transfection with pEGFP-N1 using plain-L or TATp-L as carrier. |
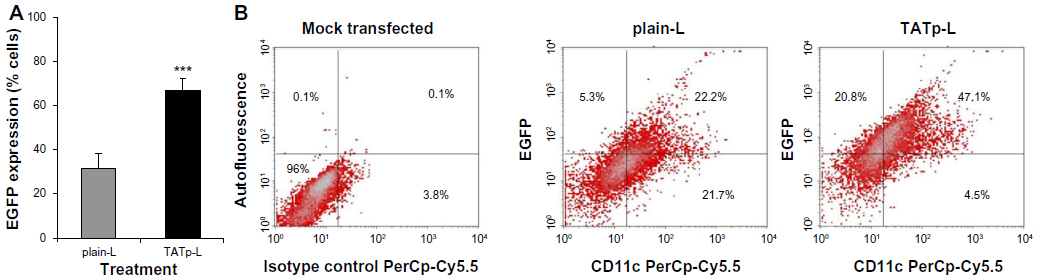 | Figure 6 EGFP transfection efficiency. |
To address the issue of the immunological activity of the de novo synthesized protein, the transfection using an encoding plasmid for the gD of BoHV-1 was performed. After a 48-hour transfection period, DC lysates were run on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by a WB against the gD. Protein was expressed only in DC treated with liposome–plasmid complexes (Figure 7A). However, the expression level achieved with the TATp-L treatment was two-fold higher than the expression level achieved with the plain-L treatment as it was determined by the ImageJ densitometric analysis of the WB bands. As a loading control, a duplicate WB was developed using a BoHV-1 polyclonal Ab that showed an unspecific band in all samples with the same intensity (Figure 7C).
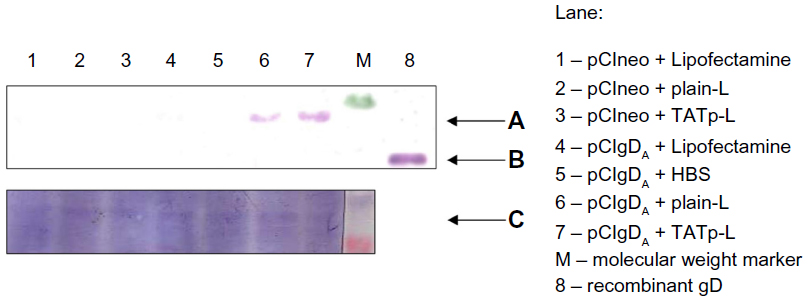 | Figure 7 Visualization of the gD protein from BoHV-1 by WB. |
Positive nanovehicle controls performed with commercial transfection reagents (plasmid plus Lipofectamine or Lipofectin) were very toxic to the DC. After 48 hours, the majority of such treated cells were dead. Negative nanovehicle controls performed with plasmid plus HBS were negative in all experiments (data not shown).
Secretion of IL-6 by DC after transfection with TATp-liposomes
After the transfection period (48 hours), supernatants of DC cultures were screened for IL-6, IL-10, IL-12, and TNFα. Only IL-6 was found in the supernatants of DC treated with TATp-L plus pCIgDA (the plasmid for BoHV-1 gD). A significant difference in IL-6 secretion (P<0.001) was observed between TATp-L and pCIgDA and the other treatments: TATp-L plus pCIneo (empty plasmid), or plain-L plus pCIneo or pCIgDA (Figure 8). There was also IL-6 secretion with Lipofectamine or Lipofectin alone (without plasmid), and with Lipofectamine and naked plasmid, both with and without the gD encoding gene, suggesting that this response was nonspecific and due to stimulation by the transfection reagent or the naked plasmid (data not shown). In addition, when liposomes were incubated with no cargo (empty liposomes), there was no cytokine detection (data not shown).
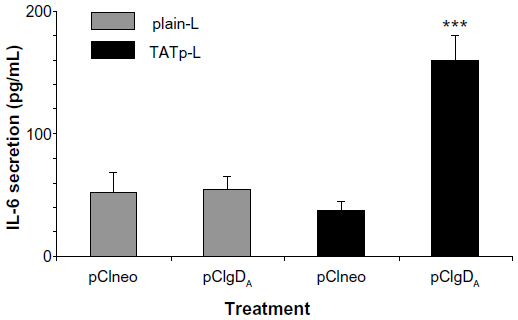 | Figure 8 IL-6 secretion by dendritic cells 48 hours post-transfection. |
Discussion
DC are especially useful for transfection in vitro and intravenous reinjection (autograft) into patients for use against cancer when a strong CTL response is required.33–37 The results presented here demonstrate how liposomal properties, as carriers for prophylactic or therapeutic vaccination, could be improved. Liposome uptake by cells using rhodamine-labeled liposomes demonstrated that liposome uptake by DC can be significantly improved with liposomes modified with TATp. This uptake phenomenon has been described for other cell types.26,38–40 It was also shown that the uptake was time-dependent since liposome accumulation in intracellular vesicles was greater at 150 minutes compared to 60 minutes. The liposomes appear to be in the cytoplasm. All pictures were taken in microchambers with living cells (in medium) so that artifacts were unlikely. In addition, as seen in Figure 4, plane B (inner section of the cell), the vesicle-like compartments containing the liposomes are very well-defined, in contrast to the surface picture, and tend to surround the nuclei without penetrating inside the nuclear membrane.
Using EGFP as a reporter gene, DC were shown to be transfected more efficiently with DNA-loaded TATp-L compared to DNA-loaded plain-L. This correlates with the liposome uptake by DC and is especially important since the ability to transfect DC is critical because they play a key role in generating an immune response. It has been demonstrated elsewhere that the low levels of DC transfection are due to the low penetration of DNA into the cytoplasm due to rapid degradation in endosomes or by specific mechanisms protecting cells against foreign DNA.41–46 To avoid degradation, DNA must both penetrate the cell membrane, be delivered directly into the cytoplasm and bypass the endocytic pathway.47 Both, plain-L and TATp-L are attracted to the DC surface because of their net positive charge. However, with short incubation times, only TATp-L significantly penetrate DC since TATp readily binds to cell surface proteoglycans and triggers receptor-mediated endocytosis (macropinocytosis).48 With sufficiently longer incubation, both formulations can be taken up by DC, evidently due to the non-receptor-mediated endocytosis. Clearly, the TATp-L transfected DC more efficiently because of their specific binding to the cell surface48 and their ability to escape from endosomes by destabilizing them.39,49–54
These experiments with EGFP demonstrate the possibility of efficient DC transfection. To prove that the expressed protein can be immunologically active, we repeated some experiments using pCIgDA instead of pEGFP. By WB, we demonstrated that DC treated either with pCIgDA-loaded plain-L or with TATp-L expressed the gD. However, transfection performed with TATp-L resulted in a protein expression two-fold higher than the expression obtained with plain-L which correlates with the result of EGFP expression. In addition, gD-transfected DC produced IL-6 specifically only when treated with pCIgDA-loaded TATp-L. IL-6 is a proinflammatory cytokine related to the maturation of B lymphocytes and their stimulation to produce antibodies, among other functions.55,56 The higher efficiency of TATp-L in gene delivery into cells, as shown by EGFP and gD expression, resulted in an amount of the de novo synthesized protein sufficient to stimulate the cytokine secretion. This was probably due to the ability of the synthesized gD to surpass the threshold needed to trigger the IL-6 secretion. It has been noted earlier by others41–43,57–62 that the toll-like receptors 2 (TLR2) and 9 (TLR9) trigger the IL-6 secretion in DC and are capable of recognizing the herpes simplex virus (HSV) envelope proteins such as gD (TLR2) and genomic HSV DNA (TLR9). BoHV-1 and HSV are highly related viruses. This suggests that the response could be specific to newly-synthesized gD that stimulates the TLR2 and/or plasmids that, in turn, stimulates the TLR9 in DC. In this case, DC transfection using TATp-L would be more efficient than plain-L in stimulating a strong humoral response by the secretion of IL-6 that subsequently enhances the B lymphocyte activity. Experiments to clarify this issue are currently underway.
There have been many in vitro gene transfer attempts using DC.4,20,63,64 APC are very resistant to transfection4,20,63–65 due to the specific mechanisms they use to take up and degrade antigens and pathogen-associated molecular patterns, including DNA, which involves various specific recognition sites including toll-like receptors.41–43
Even though methodologies based on transduction using recombinant viruses are the most widely used, they have practical limitations, such as difficulty with large-scale production and immune interference.4,20,63,64,66,67 Various non-viral transfection reagents are also available (Lipofectamine, Lipofectin, Tfx-50, Effectene®, DOTAP, FuGENE®, X-tremeGENE, CLONfectin, etc), but are very toxic to DC.20,63 We observed this with Lipofectamine or Lipofectin. By contrast, both plain-L and TATp-L were nontoxic to DC and this has also been reported for similar preparations on many other cell types.26
Our delivery system showed improved DC transfection levels when performed with TATp-L.
Even though it is very useful to have new methodologies for in vitro transfections of DC for use in “cell vaccines” or other therapies, the possibility of using them directly in vivo remains a challenging issue. However, transfections in vivo, by the injection of liposome–plasmid complexes, would have great advantages. Liposomes are excellent candidates for this purpose because of their low cytotoxicity, good stability, capacity as a carrier, and their ability to efficiently transfect DC when modified with TATp.
Acknowledgments
The authors thank Dr Lorne Babiuk for providing the anti-gD monoclonal Ab.
Disclosure
The authors report no conflicts of interest in this work.
References
Howard CJ, Charleston B, Stephens SA, Sopp P, Hope JC. The role of dendritic cells in shaping the immune response. Anim Health Res Rev. 2004;5(2):191–195. | |
Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3(12):984–993. | |
Lowrie DB, Whalen RG. DNA Vaccines. Methods and Protocols. Vol 29, 1 ed. Totowa, NJ, USA: Humana Press; 2000. | |
Landi A, Babiuk LA, van Drunen Littel-van den Hurk S. High transfection efficiency, gene expression, and viability of monocyte-derived human dendritic cells after nonviral gene transfer. J Leukoc Biol. 2007;82(4):849–860. | |
Cartier A, Masucci MG. Differential regulation of MHC class-I-restricted and unrestricted cytotoxicity by the Us3 protein kinase of herpes simplex virus-1. Scand J Immunol. Dec 2004;60(6):592–599. | |
Decman V, Freeman ML, Kinchington PR, Hendricks RL. Immune control of HSV-1 latency. Viral Immunol. 2005;18(3):466–473. | |
Deng Y, Yang D, Chen X, Chen Y. Tc1/Tc2 imbalance in the peripheral blood of patients with recurrent genital herpes. J Huazhong Univ Sci Technolog Med Sci. 2006;26(1):145–147. | |
Divito S, Cherpes TL, Hendricks RL. A triple entente: virus, neurons, and CD8+ T cells maintain HSV-1 latency. Immunol Res. 2006;36(1–3):119–126. | |
Hoshino Y, Pesnicak L, Cohen JI, Straus SE. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J Virol. 2007;81(15):8157–8164. | |
Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Curr Opin Immunol. 2004;16(4):463–469. | |
Sheridan BS, Knickelbein JE, Hendricks RL. CD8 T cells and latent herpes simplex virus type 1: keeping the peace in sensory ganglia. Expert Opin Biol Ther. 2007;7(9):1323–1331. | |
Stuart PM, Summers B, Morris JE, Morrison LA, Leib DA. CD8(+) T cells control corneal disease following ocular infection with herpes simplex virus type 1. J Gen Virol. 2004;85(Pt 7):2055–2063. | |
Verjans GM, Hintzen RQ, van Dun JM, et al. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A. 2007;104(9):3496–3501. | |
Vider-Shalit T, Fishbain V, Raffaeli S, Louzoun Y. Phase-dependent immune evasion of herpesviruses. J Virol. 2007;81(17):9536–9545. | |
Fiammenghi L, Ancarani V, Rosales T, et al. FRET microscopy autologous tumor lysate processing in mature dendritic cell vaccine therapy. J Transl Med. 2010;8:52. | |
Gholamin M, Moaven O, Farshchian M, et al. Induction of cytotoxic T lymphocytes primed with tumor RNA-loaded dendritic cells in esophageal squamous cell carcinoma: preliminary step for DC vaccine design. BMC Cancer. 2010;10:261. | |
Ma F, Zhang J, Zhang J, Zhang C. The TLR7 agonists imiquimod and gardiquimod improve DC-based immunotherapy for melanoma in mice. Cell Mol Immunol. 2010;7(5):381–388. | |
Met O, Eriksen J, Svane IM. Studies on mRNA electroporation of immature and mature dendritic cells: effects on their immunogenic potential. Mol Biotechnol. 2008;40(2):151–160. | |
Michiels A, Tuyaerts S, Bonehill A, et al. Electroporation of immature and mature dendritic cells: implications for dendritic cell-based vaccines. Gene Ther. 2005;12(9):772–782. | |
Tan PH, Beutelspacher SC, Wang YH, et al. Immunolipoplexes: an efficient, nonviral alternative for transfection of human dendritic cells with potential for clinical vaccination. Mol Ther. 2005;11(5):790–800. | |
Serafini M, Naldini L, Introna M. Molecular evidence of inefficient transduction of proliferating human B lymphocytes by VSV-pseudotyped HIV-1-derived lentivectors. Virology. 2004;325(2):413–424. | |
Gitton Y, Tibaldi L, Dupont E, Levi G, Joliot A. Efficient CPP-mediated Cre protein delivery to developing and adult CNS tissues. BMC Biotechnol. 2009;9:40. | |
Ruzza P, Calderan A, Guiotto A, Osler A, Borin G. Tat cell-penetrating peptide has the characteristics of a poly(proline) II helix in aqueous solution and in SDS micelles. J Pept Sci. 2004;10(7):423–426. | |
Levchenko TS, Rammohan R, Volodina N, Torchilin VP. Tat peptide-mediated intracellular delivery of liposomes. Methods Enzymol. 2003;372:339–349. | |
Torchilin VP, Levchenko TS. TAT-liposomes: a novel intracellular drug carrier. Curr Protein Pept Sci. 2003;4(2):133–140. | |
Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci U S A. 2003;100(4):1972–1977. | |
Pappalardo JS, Quattrocchi V, Langellotti C, et al. Improved transfection of spleen-derived antigen-presenting cells in culture using TATp-liposomes. J Control Release. 2009;134(1):41–46. | |
Zamorano P, Taboga O, Dominguez M, et al. BHV-1 DNA vaccination: effect of the adjuvant RN-205 on the modulation of the immune response in mice. Vaccine. 2002;20(21–22):2656–2664. | |
van Drunen Littel-van den Hurk S, van den Hurk JV, Gilchrist JE, Misra V, Babiuk LA. Interactions of monoclonal antibodies and bovine herpesvirus type 1 (BHV-1) glycoproteins: characterization of their biochemical and immunological properties. Virology. 1984;135(2):466–479. | |
Misra V, Gilchrist JE, Weinmaster G, Qualtiere L, Van den Hurk S, Babiuk LA. Herpesvirus-induced “early” glycoprotein: characterization and possible role in immune cytolysis. J Virol. 1982;43(3):1046–1054. | |
Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. | |
Torchilin VP, Levchenko TS, Lukyanov AN, et al. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta. 2001;1511(2):397–411. | |
Buonerba C, Ferro M, Di Lorenzo G. Sipuleucel-T for prostate cancer: the immunotherapy era has commenced. Expert Rev Anticancer Ther. 2011;11(1):25–28. | |
Di Lorenzo G, Buonerba C, Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nat Rev Clin Oncol. 2011;8(9):551–561. | |
Harada Y, Yonemitsu Y. Dramatic improvement of DC-based immunotherapy against various malignancies. Front Biosci (Landmark Ed). 2011;16:2233–2242. | |
Thara E, Dorff TB, Pinski JK, Quinn DI. Vaccine therapy with sipuleucel-T (Provenge) for prostate cancer. Maturitas. 2011;69(4):296–303. | |
Turnis ME, Rooney CM. Enhancement of dendritic cells as vaccines for cancer. Immunotherapy. 2010;2(6):847–862. | |
Marty C, Meylan C, Schott H, Ballmer-Hofer K, Schwendener RA. Enhanced heparan sulfate proteoglycan-mediated uptake of cell-penetrating peptide-modified liposomes. Cell Mol Life Sci. 2004;61(14):1785–1794. | |
Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98(15):8786–8791. | |
Tseng YL, Liu JJ, Hong RL. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol Pharmacol. 2002;62(4):864–872. | |
Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(11):823–835. | |
Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. | |
Sandor F, Buc M. Toll-like receptors. I. Structure, function and their ligands. Folia Biol (Praha). 2005;51(5):148–157. | |
Sandor F, Buc M. Toll-like receptors. III. Biological significance and impact for human medicine. Folia Biol (Praha). 2005;51(6):198–203. | |
Sandor F, Buc M. Toll-like receptors. II. Distribution and pathways involved in TLR signalling. Folia Biol (Praha). 2005;51(6):188–197. | |
Yoshikawa T, Imazu S, Gao JQ, et al. Non-methylated CpG motif packaged into fusogenic liposomes enhance antigen-specific immunity in mice. Biol Pharm Bull. 2006;29(1):105–109. | |
Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8(5):355–368. | |
Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones AT. Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans. 2007;35(Pt 4):784–787. | |
Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–315. | |
Torchilin VP. TAT peptide-modified liposomes for intracellular delivery of drugs and DNA. Cell Mol Biol Lett. 2002;7(2):265–267. | |
Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60(4–5):548–558. | |
Ruan G, Agrawal A, Marcus AI, Nie S. Imaging and tracking of tat peptide-conjugated quantum dots in living cells: new insights into nanoparticle uptake, intracellular transport, and vesicle shedding. J Am Chem Soc. 2007;129(47):14759–14766. | |
Torchilin V. Multifunctional Pharmaceutical Nanocarriers. Volume 4 of Fundamental Biomedical Technologies. New York: Springer; 2008. | |
Lundberg P, El-Andaloussi S, Sütlü T, Johansson H, Langel U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21(11):2664–2671. | |
Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol. 2005;23:1–21. | |
Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22(5):347–352. | |
Baker M, Noisakran S, Gebhardt BM, Kriesel JD, Carr DJ. The relationship between interleukin-6 and herpes simplex virus type 1: implications for behavior and immunopathology. Brain Behav Immun. 1999;13(3):201–211. | |
Geisel J, Kahl F, Müller M, et al. IL-6 and maturation govern TLR2 and TLR4 induced TLR agonist tolerance and cross-tolerance in dendritic cells. J Immunol. 2007;179(9):5811–5818. | |
Jang S, Uematsu S, Akira S, Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J Immunol. 2004;173(5):3392–3397. | |
Loures FV, Pina A, Felonato M, Calich VL. TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J Immunol. 2009;183(2):1279–1290. | |
Salio M, Cella M, Suter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol. 1999;29(10):3245–3253. | |
Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A. 2006;103(46):17343–17348. | |
Irvine AS, Trinder PK, Laughton DL, et al. Efficient nonviral transfection of dendritic cells and their use for in vivo immunization. Nat Biotechnol. 2000;18(12):1273–1278. | |
Tan PH, Beutelspacher SC, Xue SA, et al. Modulation of human dendritic-cell function following transduction with viral vectors: implications for gene therapy. Blood. 2005;105(10):3824–3832. | |
Tan PH, Yates JB, Xue SA, et al. Creation of tolerogenic human dendritic cells via intracellular CTLA4: a novel strategy with potential in clinical immunosuppression. Blood. 2005;106(9):2936–2943. | |
Jonuleit H, Tüting T, Steitz J, et al. Efficient transduction of mature CD83+ dendritic cells using recombinant adenovirus suppressed T cell stimulatory capacity. Gene Ther. 2000;7(3):249–254. | |
Parker AL, Newman C, Briggs S, Seymour L, Sheridan PJ. Nonviral gene delivery: techniques and implications for molecular medicine. Expert Rev Mol Med. 2003;5(22):1–15. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.