")
Back to Archived Journals » Current Biomarker Findings » Volume 5
Idiopathic pulmonary fibrosis biomarkers: clinical utility and a way of understanding disease pathogenesis
Authors Flynn M, Baker E, Kass DJ
Received 22 January 2015
Accepted for publication 25 February 2015
Published 14 May 2015 Volume 2015:5 Pages 21—33
DOI https://doi.org/10.2147/CBF.S81362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Hung Khong
Matthew Flynn, Elisabeth S Baker, Daniel J Kass
Dorothy P and Richard P Simmons Center for Interstitial Lung Disease, Division of Pulmonary, Allergy, and Critical Care Medicine, University of Pittsburgh, Pittsburgh, PA, USA
Abstract: Idiopathic pulmonary fibrosis (IPF) is a typically fatal disease that remains incompletely understood despite intense study and the arrival of drugs that may alter the natural history of the disease. Rendering an accurate diagnosis and predicting prognosis remain challenging problems to clinicians. One potential solution to these clinical problems is the identification of IPF biomarkers, easily measured factors that can be employed to predict clinical behavior. Candidate biomarkers have been identified by research in the laboratory on potential culprit cells or genes that may contribute to the pathogenesis of IPF. In this review, we present the current data on a number of well-studied IPF biomarker candidates and their potential role in the pathogenesis of disease. We also establish a framework for evaluating utility of incorporating these IPF biomarkers into clinical practice.
Keywords: idiopathic pulmonary fibrosis, usual interstitial pneumonia, biomarker, matrix metalloproteinases
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive scarring of the lung parenchyma characterized histologically by the usual interstitial pneumonia (UIP) pattern – a disease limited to the lungs and lacking a well-defined etiology.1 IPF has remained a rare disease, occurring predominantly in older men. Recent epidemiologic studies suggest an increasing incidence, possibly as high as 94 per 100,000, using refined definitions of disease.2 The mechanism of disease remains unclear. Recent theories have moved away from the long-standing paradigm of chronic inflammation to one that favors recurrent alveolar epithelial cell injury and aberrant repair.3 The interplay of pro-fibrotic cell types and the mediators associated with their recruitment, proliferation, and differentiation is presently the subject of intensive research.4 Another elusive feature of IPF is its heterogeneous natural history: many patients demonstrate a stable or slow decline, while for others, the decline is precipitous or fraught with the so-called acute exacerbations, sudden deteriorations of pulmonary function of unclear etiology.5 Regardless, overall prognosis is quite dismal, with a median survival of 3.8 years from diagnosis.2 For decades, the only US Food and Drug Administration-approved treatment for this deadly disease has been lung transplantation, but with the approval of pirfenidone and nintedanib to treat IPF,6,7 the IPF landscape is sure to change.
The clinical problem
When evaluating a patient with pulmonary fibrosis, the clinician is faced with the following questions:
- Is this disease IPF? There are several diseases that may present clinical, radiographic, and pathologic manifestations that are quite similar to IPF. Making an accurate clinical diagnosis is critical as these diseases may be treated differently. Even multidisciplinary experts demonstrate significant interobserver disagreement in IPF diagnosis based upon high-resolution computed tomography (CT) and histologic review when blinded.8
- If it is IPF, will this patient benefit from the newly available treatments? At an estimated cost upwards of $100,000 per year for a course of treatment with pirfenidone or nintedanib,9 predicting a priori who is likely or unlikely to benefit is critical.10
- Who will experience a more progressive course of disease? In practice, IPF patients tend to be referred for transplantation evaluation early on in the course of their disease – an international consensus report from 2006 outlined a guideline for referral in any patient with radiographic or histologic evidence of UIP.11 But identifying who should be prioritized for early transplant from those who will follow a more indolent course is currently one of the most critically important but unanswered questions for IPF clinicians.
Biomarkers appear to be the answer to these important clinical questions.
What is a biomarker?
Before exploring the current literature on IPF-specific biomarkers, it is beneficial first to consider the subject of biomarkers more generally and to create a framework for evaluating their potential utility. As defined by a National Institutes of Health expert-working group, a biomarker is “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention”.12 As it stands, a biomarker denotes many things, including vital signs, general chemistry panels, specialized biochemical analyses, physiologic testing, and radiographic studies. While all of these measurements may have a role in the care of IPF patients, we focus this review on molecular and cellular biomarkers. Molecular and cellular biomarkers encompass any measurable element from the lung via exhaled condensate or bronchoalveolar lavage fluid (BALF) as well as urine, blood, or saliva. Technological advances in next-generation sequencing techniques have greatly expanded the armamentarium of potential biomarkers, but to date, there are no measured markers that have been implemented clinically except in Japan.13 A biomarker with clinical utility requires a sufficient measure of accuracy and reproducibility – that is, one would like a biomarker to have strong performance characteristics, including high sensitivity and specificity, and strong positive or negative predictive values and likelihood ratios, depending on specific clinical usage. Additional factors that are equally as important include safety and ease of obtaining the sample, acceptable costs to perform the test, and reasonable availability of testing. A schematic workflow to translate a candidate biomarker into a clinical test is presented in Figure 1. The potential for molecular biomarkers in IPF is significant, and research suggests their possible value in confirming an accurate diagnosis of IPF, determining genetic predisposition for the development of IPF, providing prognostic information regarding disease course and mortality, measuring disease progression and activity, and gauging response to potential therapies.
 | Figure 1 Translation of a candidate biomarker into clinical practice. |
What do candidate biomarkers tell us about IPF pathogenesis?
In their seminal review of IPF, published in 2001, Selman et al very elegantly refuted the commonly held notion of inflammation as the prominent causative factor of IPF.3 The studies of the last 14 years since this publication were some of the most critically important studies of IPF. Many genes and pathways of the deranged epithelial phenotype in IPF were identified that led to the study of many of the epithelial cell-derived candidate biomarkers described below. Pathologic imbalance of proteases and antiprotease studies also lead to candidate biomarkers, particularly the matrix metalloproteinases (MMPs), described in the Fibrogenesis/matrix remodeling section. What perhaps has been most unexpected was the recent identification of genetic factors such as Mucin 5B (MUC5B) polymorphism or the resurgence of inflammation as a critical component of the fibrotic process. In their updated review of IPF in 2011,3 King et al note that pulmonary fibrosis remains a disease of aberrant healing but that inflammation may play a role in the progression of disease in a subset of patients.3 This shift in thinking, we suggest, is reflected in part by the relative increase in the number of inflammatory candidate biomarkers including semaphorin 7a (Sema7a), BLyS, and chemokine (C-X-C motif) ligand 13 (CXCL13) compared to epithelial cell-derived biomarkers. This new focus of IPF biomarker research may also reflect the shift away from lung tissue-derived biomarkers to the less invasive peripheral blood biomarkers.14 Therefore, we suggest that candidate biomarkers speak to prevailing theories of the pathogenesis of IPF, and as these theories have evolved over time, so have the biomarkers. We have grouped the biomarkers below into categories based on their purported site of action. A similar categorization of IPF biomarkers has recently been published.15 The list below is roughly chronological. We review the clinical data, and we also discuss these biomarkers within the context of the larger shifts in our understanding of IPF. We first focus on biomarkers derived from epithelial cell injury, followed by a discussion of extracellular signaling molecules, matrix remodeling, cellular markers, and finally inflammatory biomarkers. Table 1 summarizes the data.
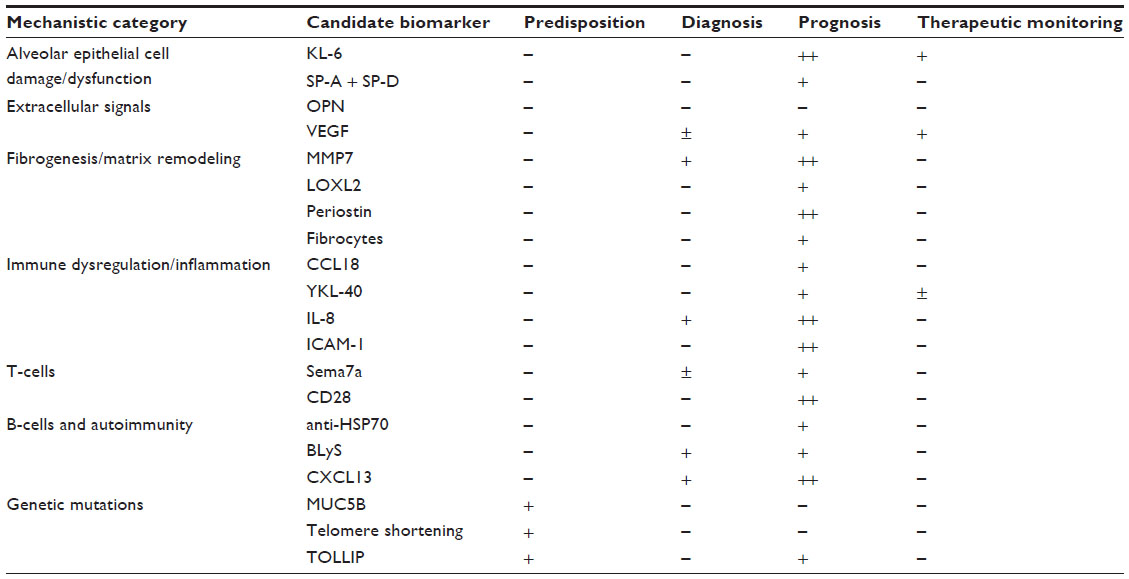 | Table 1 Summary of candidate biomarker data |
Alveolar epithelial cell damage/dysfunction
Krebs von den Lungen 6
Krebs von den Lungen 6 (KL-6) antigen is a transmembrane mucin, or glycoprotein, encoded by MUC1 (Mucin 1, cell surface associated) that is expressed on the apical surface of type II alveolar epithelial cells. The precise function is unknown, but studies suggest that KL-6 has chemotactic activity on fibroblasts as well as anti-apoptotic effects on stimulated fibroblasts via interaction through an unknown receptor.16 KL-6 is in clinical practice in Japan, and several studies have been completed in this population. While KL-6 level elevation may be sensitive for interstitial lung disease (ILD), with differentiation demonstrated between ILD and controls, it is not specific for IPF. It is elevated in numerous ILD conditions, without significant differences between the UIP and nonspecific interstitial pneumonia (NSIP) histologies; therefore, it appears to have limited utility in diagnosis.17,18 This biomarker has shown promise in prognostication: in a smaller study with 27 patients, the authors found that lower baseline levels of KL-6 at time of diagnosis, <1,000 U/mL, have a more favorable 3-year survival rate.19 In a larger scale study of 219 patients with ILD, of which 183 were determined to have UIP pattern on high-resolution CT, it was found that patients with lower baseline KL-6 levels had significantly improved mortality over a 6-year period. The cut-off used in this study was again 1,000 U/mL, and, notably, those subjects with markedly elevated KL-6 levels had particularly poor survival.20 An additional study subsequently determined that baseline KL-6 levels above 1,300 U/mL were sensitive predictors for higher incidence of experiencing an acute exacerbation of IPF over a 3-year follow-up period, making it a potentially valuable tool for prognostication.21
While the approved therapies for IPF are new, with limited data available to date, there is some evidence that KL-6 may be a marker for response to drug therapy. A small study of 14 patients with acute exacerbations of IPF examined KL-6 levels pre-pulse dose steroid administration as well as 1 week and 3 weeks post-administration: patients who survived 3 months had decreasing levels of KL-6 at each follow-up interval, which could suggest that KL-6 levels fall either in response to therapy or that elevated KL-6 levels reflect poorer prognosis.22 Finally, although not a primary end point, a recent study investigating the use of pirfenidone found that the medication not only attenuated a decline in pulmonary function but also reduced serum levels of KL-6, again suggesting the biomarker as an indicator of therapeutic response.23 Probably, the most significant limitation to a wider application of the results of most KL-6 studies is the limited population of the studies, although a study including German subjects looking at MUC1 polymorphisms suggests that extrapolation may be possible.13 As a molecular biomarker in IPF, KL-6 seems to demonstrate promise primarily in prognostication, with some data hinting at possibilities for therapeutic response monitoring.
Surfactant proteins A and D
Surfactant proteins (SPs) are lipoprotein complexes synthesized and secreted by type II alveolar epithelial cells with identified roles in reducing the surface tension at the lung air–liquid interface and promoting lung host defense. SFTPA (SP-A) and SFTPD (SP-D), specifically, have demonstrated roles in the innate immune system through direct binding to microorganisms and regulation of macrophages, and also in the adaptive immune system through interacting with both dendritic cells and T-cells. Beyond protecting the host, evidence also points to a role in modulating the inflammatory response to protect the lung from injury.24,25 One suggested mechanism is that chronic type II cell injury leads to cell turnover, possible somatic DNA mutations, resulting in apoptosis via endoplasmic reticulum stress secondary to protein misfolding or telomerase shortening. Subsequently, through debated pathways, epithelial cell injury results in fibrosis.25 Strengthening the case, limited studies have linked mutations in SP-A with familial IPF; although given sparse available data to date, its potential use in screening at-risk populations cannot be determined.26 Mutations in SP-C (SFTPC) have clearly been associated with familial pulmonary fibrosis.27–30
SPs have been measured in both serum and BALF samples, with results suggesting possible roles in diagnosis and prognostication. In a 2002 study referenced above, 33 patients with ILD, including 21 diagnosed with IPF, were found to have serum levels of SP-A and SP-D that were significantly elevated compared to controls; significant differences between IPF and other ILD subjects were observed only with SP-D.17 A small study evaluating levels of SP-A in BALF samples of IPF and sarcoidosis patients suggests that lower levels of SP-A are present in IPF patients.31 This is similar to findings in a prior study from 1995 that measured SP-A in the BALF of IPF patients versus healthy subjects, with results significant for lower SP-A levels in IPF, as well as lower levels equating to diminished survival.32 However, when compared to other ILD patients in a separate study, a reduction in BALF SP-A content was not found to be specific for IPF.33 Further contradicting those prior studies, a more recent evaluation of BALF in patients with IPF and hypersensitivity pneumonitis showed increased levels of SP-A in hypersensitivity pneumonitis (HP) compared to control samples.34
Serum testing has yielded mixed results: a large study of IPF patients in 2002 found limited utility in SP serum levels in distinguishing IPF from other types of ILD, but it did find elevated levels of both SP-A and SP-D to correlate with poor survival in IPF patients.35 Subsequently, a study of 82 IPF patients determined that elevated SP-A levels at initial diagnosis are significantly associated with 1-year mortality or transplant, while SP-D was not (although it did have a similar trend).36 Meanwhile, results looking only at the value of SP-D as a marker of prognosis did find significantly increased mortality in IPF patients with elevated levels.37 As a biomarker, SPs appear to have limited clinical utility in the differentiation of IPF from other ILDs, with more encouraging results in the field of prognosis.38
Extracellular signals
Osteopontin
Osteopontin (OPN) is a glycosylated phosphoprotein that is expressed in many tissues throughout the body, and within IPF lungs, it is localized to alveolar epithelial cells and macrophages.39 Significant advances in the understanding of this marker were made by Pardo et al, who discovered OPN to be one of the most highly upregulated genes in IPF via microarray gene expression studies on IPF lung tissue.39 In addition to the aforementioned immunohistochemical results, this group also found significantly elevated levels of OPN in BALF samples of 18 IPF patients compared to controls. They also demonstrated upregulation of, and co-localization with, another biomarker MMP7, and demonstrated in vitro associations of OPN with human fibroblast and epithelial cell growth and migration.39 A small study of 17 ILD patients and nine patients with sarcoidosis found significantly elevated levels of serum OPN in both conditions compared to controls; there was also a significant difference found between ILD and sarcoidosis patients, although IPF patients were statistically indistinguishable from other ILD conditions.40 This study noted an inverse correlation with arterial partial pressure of oxygen (PaO2) levels but not with pulmonary function test (PFT) values. There was no significant correlation between OPN and other biomarkers, including KL-6 and SP-A and SP-D.40 Despite the above discoveries strongly supporting a role for OPN in the pathogenesis of IPF, the data are insufficient to support its use as a biomarker for IPF.
Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF) is a signaling protein that functions via tyrosine kinase receptors to modulate various angiogenic processes, including inhibition of endothelial cell apoptosis, regulation of vascular permeability, and proliferation, migration, and differentiation of endothelial cells.41 This protein is highly expressed in alveolar epithelial cells, as well as lung macrophages, smooth muscle, and myofibroblasts.42 A number of VEGF isoforms exist, the first discovered and most well known being VEGF-A; however, most IPF literature does not differentiate, and discussions refer to pan-VEGF isoforms unless specifically noted.43 A study by Meyer et al measuring VEGF levels in BALF and serum found significantly depressed values in BALF of IPF patients compared to controls and sarcoidosis and cystic fibrosis patients; serum levels did not demonstrate any significant differences.44 A few years later, another investigation of BALF by Koyama et al determined that VEGF concentrations were significantly reduced in all chronic pulmonary disease states evaluated (IPF, ILD related to connective tissue disease, and sarcoidosis), as well as in healthy smokers.45 Serum VEGF levels were reevaluated in a 2010 study that included 41 IPF patients, with results identifying significantly elevated VEGF levels in IPF patients with high Alveolar–arterial (A–a) oxygen gradients compared to IPF patients with lower A–a gradients or healthy controls. There was an additional finding of baseline serum VEGF values inversely correlating with subsequent forced vital capacity (FVC) declines.46 A very recent study of serum VEGF-A plasma levels found significantly elevated levels in IPF patients compared with controls, without any correlation with PFTs.47 As a biomarker, VEGF BALF levels appear significantly associated with IPF and could possibly be a component of evaluation in patients with an uncertain diagnosis, but it is probably of limited utility. Serum levels are more practical as a biomarker, and may have some utility in a diagnostic algorithm, as well as prognosis of clinical course. Nintedanib, one of the two newly approved therapies for IPF, is a tyrosine kinase inhibitor that targets VEGF signaling along with other pathways.7 Phase III trials have shown that treatment slows the decline of pulmonary function. Given the possibility that VEGF signaling is a part of pulmonary fibrosis, more study is necessary to determine how VEGF will perform as a biomarker of treatment success.
Fibrogenesis/matrix remodeling
Matrix metalloproteinases
MMPs are a collection of zinc-dependent endopeptidases that are collectively involved in the remodeling of the extracellular matrix and are thought to have roles in both normal organogenesis and inflammation. In fibrotic lung disease, they are purported to have a role in the observed dysregulated collagen turnover.48 MMP7 has been particularly well studied, with gene and protein expression levels found to be significantly increased in fibrotic lung, localizing to alveolar and bronchiolar epithelial cells. Furthermore, in vivo, MMP7 knockout mice were protected from bleomycin-induced fibrosis.49 An additional study found active MMP7 to immunolocalize to alveolar macrophages and hyperplastic epithelial cells in IPF lungs.50
A study aiming to evaluate MMP levels in 20 IPF patients determined that MMP3, MMP7, MMP8, and MMP9 were all elevated in BALF compared to healthy controls. The authors also found that all four of these MMPs were more elevated in those with poorer prognoses, and in particular, MMP8 and MMP9 were also significantly associated with rapidly declining pulmonary function as defined by persistent FVC reduction >10% over a 1-year period.51 A recent evaluation of MMP8 in IPF determined that there were increased BALF and serum levels compared to controls, although these increased levels were not found to correlate with declining lung function or mortality.52 Further studies of MMP7 in IPF patients have demonstrated increased BALF levels above normal controls but nonsignificant differences in serum levels and lack of differentiation between IPF and other ILDs.50,53,54 An analysis of multiple plasma proteins in IPF versus a control population that included chronic obstructive pulmonary disease (COPD), sarcoidosis, and HP identified MMP1 and MMP7 as primarily overexpressed proteins in IPF. In combination, MMP1 and MMP7 significantly differentiated IPF patients from the other chronic pulmonary conditions; interestingly, MMP7 levels correlated with subclinical ILD and were negatively associated with PFT results, suggesting possible roles in early diagnosis and gauging disease severity.55 Finally, a recent large study incorporating 241 IPF patients, with both derivation and validation cohorts, analyzed the concentrations of 92 serum proteins and found five proteins, including MMP7, to be associated with prognosis and mortality.56 What is perhaps most notable is that the robust nature of the results led the investigators to derive a severity prediction rule based on MMP7 levels. Overall, MMP7 appears to have the greatest individual clinical utility as a biomarker, primarily in the realm of prognosis, especially when combined with other biomarkers. MMP7 may have limited utility, again in combination with other biomarkers, in differentiation of IPF from other ILDs. The suggestion that MMP7 may have a role in diagnosis of subclinical disease is particularly intriguing, although further investigation is necessary.
Lysyl oxidase-like 2 (LOXL2)
Inhibition of LOXL2 in IPF is currently being studied in a clinical trial by Gilead Biosciences. Researchers from Gilead in collaboration with university-based coinvestigators found that higher serum levels of LOXL2 correlated with IPF disease progression.57 The authors note that implementation of LOXL2 as a measured biomarker will require validation in larger cohorts.
Periostin
Periostin is an extracellular matrix protein, part of a class of “matricellular” proteins that modulate cell–matrix interactions via signaling mechanisms and have identified roles in cardiac development and remodeling, tumorigenesis, and wound repair.58–60 The mechanism of action is not well defined in IPF, although a murine model has suggested a role in induction of chemokines and recruitment of neutrophils and macrophages.61 A study of 91 ILD patients, 51 with IPF, used immunohistochemistry to demonstrate strong expression of periostin in fibroblastic foci of lung tissue in UIP and NSIP. Further work by this group noted significantly elevated serum levels in IPF and NSIP compared to cryptogenic organizing pneumonia (COP) and normal controls, as well as inverse correlation with 6-month measurements of pulmonary function, suggesting a role for the biomarker in prognostication.62 The findings were largely verified by a second study of 54 IPF patients, with immunohistochemistry demonstrating increased staining for periostin in fibrotic lung tissue, as well as increased production of periostin messenger RNA by IPF fibroblasts. Clinically, the group was able to demonstrate correlation with serum periostin levels and disease progression, as defined by death, acute exacerbation of IPF, transplant, reduction of FVC by >10%, or decline of diffusing capacity of the lung for carbon monoxide (DLCO) by >15% at 48-week follow-up.63
Circulating fibrocytes
In 2004, two studies published nearly simultaneously64,65 identified a bone marrow-derived collagen-producing cell, termed “fibrocyte”,66 as a critical player in matrix production in experimental lung fibrosis. Since then, multiple studies have examined these cells, defined as expressing both the common leukocyte antigen CD45 and collagen I, when isolated from the peripheral blood65 as biomarkers for disease severity in IPF and in other non-IPF diseases.67–70 The fibrocyte bridges our categories of matrix and remodeling and immune dysregulation as it exhibits properties of both. “Bursts” of fibrocytes did not correlate with death in a study of Hermansky–Pudlak syndrome,67 suggesting that deterioration or death does not correlate with absolute fibrocyte numbers and potentially limits enthusiasm for the fibrocyte as a biomarker.71 Further study will be necessary to assess absolute numbers of fibrocytes, their behavior over time, and their relationship with pulmonary function.
Immune dysregulation/inflammation
Chemokine (C-C motif) ligand 18
Chemokine (C-C motif) ligand 18 (CCL18), also known as pulmonary and activation-regulated chemokine, is an extracellular signal primarily expressed by alveolar macrophages that recruit lymphocytes and dendritic cells to foci of inflammation. Experimental studies indicate the existence of a positive feedback loop whereby alveolar macrophages are stimulated by native collagen to produce CCL18 and fibroblasts respond to CCL18 by synthesizing collagen.72,73 In a study of 55 patients with ILDs by Prasse et al, CCL18 levels were found to be elevated in both BALF and serum, most impressively in IPF, NSIP, and scleroderma. Moreover, elevated levels of CCL18 correlate with worse PFT.74 A further prospective study of 72 IPF patients, using a cut-off value for serum CCL18 concentrations of 150 ng/mL, found that patients above this level have significantly decreased measures of pulmonary function at 6-month follow-up, as well as increased mortality.75 A more recent investigation of CCL18 levels in the serum, BALF, and alveolar macrophage culture supernatants of patients with a variety of ILDs found elevated levels in various pulmonary conditions, most significantly in HP. This study posited that CCL18 may therefore reflect inflammatory response.76 Presently, collective results suggest limited utility for diagnostic purposes of this biomarker; however, the correlation with PFT trends, a known marker of disease progression, suggests utility as a prognostic tool in IPF patients.
YKL-40
A chitinase-like glycoprotein (chitinase 3-like 1, CHI3L1 [cartilage glycoprotein-39]), studied in the literature as YKL-40, has previously been associated with fibrosis in liver disease and to correlate with the degree of airway remodeling in severe asthma.77,78 While the mechanism of YKL-40 is not well understood, research has suggested roles in regulating connective tissue cell proliferation and angiogenesis.79,80 Within the lung, immunohistochemistry has demonstrated enhanced expression of YKL-40 in airway epithelial cells and alveolar macrophages.81 In a study of 63 IPF patients, Furuhashi et al found elevated levels of YKL-40 in serum and BALF samples, as well as a correlation with DLCO and KL-6 levels (serum YKL-40 only), suggesting a possible role for the biomarker in gauging disease activity and prognosis.81 Further work by Korthagen et al again determined that serum and BALF samples in 85 IPF patients are significantly elevated compared to controls; however, they found no correlation between serum levels and BALF levels or with PFT results. They followed the patients for >4 years and found that combining serum and BALF levels of YKL-40 allowed for stratification by mortality, with those patients having higher levels in both serum and BALF demonstrating increased mortality.82 Finally, a recent study of 315 patients with various ILDs, including 185 with IPF, determined significant elevations of YKL-40 in all ILDs compared to the control population, with the highest levels noted in NSIP, IPF, and COP.83 While the biomarker does not appear to have any role in diagnosis, a subset of the study with available serial follow-up did find levels of YKL-40 decreased over time in COP, a disease which tends to respond to available therapies, while remaining consistently elevated in IPF patients, suggesting a possible role in evaluating disease activity and therapeutic monitoring.83
Interleukin-8
Interleukin-8 (IL-8; also known as CXCL8) is a cytokine produced by phagocytes exposed to inflammatory stimuli and serves as a chemoattractant for neutrophils.84 A study of the BALF in patients with IPF and fibrosis associated with known rheumatologic disease found significantly greater concentrations of IL-8 and neutrophils in both patient populations compared to healthy controls or those with non-pulmonary rheumatologic disease, suggesting that IL-8-mediated neutrophil chemotaxis is mechanistically involved in pulmonary fibrosis. When compared directly, the IPF population had significantly greater levels of IL-8 than those with systemic rheumatologic disease.85 A similar study of BALF samples from IPF patients attempted to correlate IL-8 levels, neutrophilia, and radiographic fibrosis with results. The authors found that IL-8 and neutrophils levels were significantly elevated in IPF compared to controls. IL-8 levels did not, however, appear to correspond to radiographic abnormalities on high-resolution CT imaging.86 In 1998, a study was performed specifically to evaluate whether IL-8 serum levels could serve as a biomarker. This study by Ziegenhagen et al evaluated IL-8 levels in the serum and BALF of IPF patients and found that IL-8 serum levels were significantly elevated in the IPF population compared to controls. Furthermore, the IL-8 serum levels positively correlated to elevated BALF neutrophils and BALF IL-8 levels. This study then applied known clinical data and noted a negative correlation in lung function testing and PaO2 levels in patients with elevated serum IL-8 levels, suggesting a possible role for serum IL-8 as a tool for prognostication or monitoring of disease status.87 These data are bolstered by a more recent investigation of the serum levels of several cytokines in IPF patients that found cytokines IL-2, IL-8, IL-10, and IL-12 to be significantly elevated, with IL-8 specifically negatively correlating with PFTs.88 While studying a broader ILD population, and including only a small sample of IPF patients, a study by Vasakova et al similarly discovered a negative correlation between IL-8 BALF levels and lung function testing.89 Finally, in a study by Richards et al referred to elsewhere in this review, high concentrations of plasma IL-8 reflected poor overall survival, transplant-free survival, and disease progression-free survival in the derivation cohort. Elevated IL-8 was also predictive of overall survival and poor transplant-free survival in an independent validation cohort.56 Several investigations certainly appear to support a role for IL-8 as a biomarker for prognostication given replicated relations with PFT declines, as well as the validated associations with survival.
Intercellular adhesion molecule-1
Intercellular adhesion molecule-1 (ICAM-1) is a transmembrane glycoprotein expressed on leukocytes and endothelial cells, with roles in T-cell-mediated cell defense and inflammation, participating in activated leukocyte migration.90 In 1992, Shijubo et al evaluated the serum levels of ICAM-1 in a variety of patient populations, including IPF, non-IPF ILD, pneumonia, and non-pulmonary inflammatory disorders, with a finding that levels are significantly elevated in IPF compared to other conditions. Additional immunohistochemistry analysis on lung tissue from the IPF patients demonstrated expression of ICAM-1 on alveolar epithelial cells.91 A follow-up study investigated the serum and BALF levels of ICAM-1 in IPF patients and again found significant elevations in the serum of IPF patients compared to both sarcoidosis patients and healthy volunteers, although BALF levels in IPF patients were not significantly elevated.92 In another study of the serum levels of several ICAM proteins in IPF, the authors found both ICAM-1 and ICAM-2 to be significantly elevated in IPF patients and secondary ILD patients. The group then reviewed PFT results and found that while ICAM-2 was significantly inversely associated with DLCO levels, ICAM-1 levels were not significantly associated with any particular PFT.93 Recently, this peripheral blood protein was also implicated in disease outcome predictions, including overall mortality, transplant-free survival, and even progression-free survival in the previously referred to study by Richards et al.56 Overall, early studies of this biomarker provide support for ICAM-1 having a plausible role in IPF pathogenesis, although its uses as even a diagnostic tool appear limited given similarly elevated levels in various ILD conditions. The recent larger study by Richards et al, however, strongly suggests at least a role in prognostication.56
T-cells: Sema7a and CD28
Sema7a is a membrane-bound protein that is part of a family of proteins initially identified as having a role in axonal formation in embryonic development.94 Individually, Sema7a, which is expressed on activated T-cells, has identified functions in T-cell-mediated inflammation and immune regulation, and has been demonstrated to be a necessary component of transforming-growth factor beta 1-mediated pulmonary fibrosis in animal models.95–97 A study by Reilkoff et al demonstrated that Sema7a expression localizes to macrophages and CD4+ lymphocytes in IPF lungs and is expressed on peripheral CD4+ cells in IPF serum, specifically T-regulatory cells (Tregs). Furthermore, the study demonstrates significantly increased expression of Sema7a+ Tregs in IPF subjects compared to a control population, and in follow-up, noted the highest increases in patients with progressive IPF, suggesting a role in prognostication.98 Treg participation in IPF has a somewhat conflicting history, however, with studies demonstrating both protective and causative roles in fibrosis, suggesting that further research on this promising subset of Tregs is warranted.99–102
Markedly decreased expression of CD28, a necessary component of T-cell activation, on the surface of CD4+ T-cells was correlated with a more severe 1-year outcome in IPF.103 These findings were supported further by genomic analysis of peripheral blood mononuclear cells from two separate IPF cohorts that showed that low expression of four genes, LCK, ITK, ICOS, and CD28, was associated with increased IPF mortality.104 These four genes all map to the T-cell co-stimulatory signal pathway and suggest that repeated cycles of T-cell stimulation are associated with the development of a pathologic T-cell phenotype and a significantly worsened prognosis in IPF.103
B-cell pathways: autoantibodies, BLyS, and CXCL13
In what appears to be a return to some of the earliest work on the pathogenesis of IPF,105,106 several recent studies have focused directly on autoantibodies and B-cells, using BLyS and CXCL13 as markers of B-cell activation. Recent data implicate B-cell pathways in the pathogenesis of IPF. Isolation of autoantibodies to HSP70 in patients with IPF was associated with a poorer prognosis.107 BLyS, also known as B-cell-activating factor (BAFF, official gene name TNFSF13b, tumor necrosis factor (ligand) superfamily, member 13b), is a cytokine in the tumor necrosis factor family that is secreted by lymphocytes and is critical to B-cell maturation and antibody production. Xue et al found that plasma BLyS was significantly elevated in patients with IPF when compared to controls without lung disease and with COPD.108 Furthermore, there was a significant correlation between elevated pulmonary artery pressures and BLyS levels in patients with IPF, as well as lung function, as measured by FVC. Post hoc analyses from this study demonstrated that IPF patients with the most elevated BLyS levels (highest 25%) had increased 1-year mortality.
Another biomarker of interest is CXCL13, an agent that promotes B-cell trafficking to immunologic foci and has also been implicated in autoimmunity.109 DePianto et al looked at lung tissue from patients with IPF and controls and correlated distinct histological features (lymphoid aggregates) of the IPF lung with elevated CXCL13 expression, by immunohistochemistry, plasma levels, and transcriptomics.109 Similar to BLyS described above, elevated CXCL13 levels correlated negatively with both lung function, measured by DLCO, and survival in patients with IPF. Vuga et al measured CXCL13 expression in both lung tissue and plasma in patients with IPF and COPD controls and found significantly increased CXCL13 levels in the IPF population, which correlated negatively with survival and pulmonary function as measured by DLCO.110
Genetic mutations
As discussed, biomarkers are not limited to the measure of protein levels in peripheral blood or BALF, and recent genetic investigations of IPF patients have yielded a number of intriguing variations that may hold clinical utility. In 2011, Seibold et al identified a common polymorphism in the promoter region of MUC5B, a gel-forming mucin gene expressed by bronchial epithelial cells, while performing a genome-wide scan of 82 families with familial interstitial pneumonia (FIP). When applied to a population including FIP, IPF, and control patients, the mutation was identified in >30% of FIP and IPF patients but only 9% of controls.111 This group also demonstrated that the mutation is associated with increased gene expression, although the exact mechanism of pathogenesis remains unclear.111 In support of the data from Seibold, Zhang et al confirmed this association in a larger case–control study including 341 IPF patients and 802 controls.112 While the MUC5B polymorphism is strongly associated with IPF, a retrospective analysis found improved survival in IPF patients with the polymorphism compared to those without, a finding that leads to speculation of differing phenotypes in those with the mutation.113 Mutations in genes encoding telomerase components (TERT/TERC) that lead to telomere shortening also appear associated with IPF.114,115 Even in the absence of known telomere-shortening mutations that were initially discovered in evaluations of familial IPF patients, significantly more patients with sporadic IPF have been noted to have circulating leukocytes with short telomeres compared to controls.116 Similar findings by Alder et al found that nonfamilial IPF patients, even in the absence of familial mutations, have shorter peripheral blood leukocyte telomeres than age-matched controls.117 Finally, a more recent genome-wide association study identified a common single-nucleotide polymorphism in TOLLIP, a toll-interacting protein felt to play a role in innate immune system regulation, that correlates with increased mortality.118 Clearly, these biomarkers have tantalizing utility in early prognostication and perhaps even identifying patients at risk for development of IPF. Similar studies have been identifying additional genetic variants that, with further investigation, will likely broaden the genetic understanding of IPF and possibly serve similar functions as biomarkers.
Future directions
We hope that we have made a convincing argument that the study of biomarkers is an essential part of IPF research. We suggest that biomarkers have the potential to impact all aspects of the clinical care of IPF patients including diagnosis and prognosis. There is a need for biomarkers obtained by less invasive techniques. With literature suggesting that there is increased short-term mortality following lung biopsy as well as possible increased risk of acute exacerbation,119–121 it is likely that there will be fewer and fewer lung biopsies in the future. The most recent consensus guidelines even dissuade from routine bronchoscopy for cellular analysis in the majority of patients.1 We are primarily left with peripheral blood sampling, with most studies to date focusing on measurable levels of peripheral blood proteins. Newer studies will likely focus on the data that are obtainable from peripheral blood mononuclear cells.104 Another potential source of information may be the isolation of circulating microRNA species.122,123
How should biomarkers be studied? Hypothesis-generating omics experiments will be critical in this regard. The first step already exists. The Lung Genomics Research Consortium (LGRC) was funded to be the largest research database of clinical and genomics information in lung disease.11 This public database represents transcriptomic analysis of lung tissues from patients with IPF combined with cross-sectional clinical data. There are several shortcomings, however, with the LGRC data. First, for the study of biomarkers, as we have noted above, we must be able to derive data from more accessible sources such as blood. Second, because these data are cross-sectional, we do not see how these biomarkers behave over time. Therefore, we suggest that biomarker studies must accompany every clinical trial in IPF.14 Clinical trial participants represent a large and “captive” audience. For example, the ASCEND trial sponsored by Intermune studied 555 patients worldwide.6 Every clinical trial visit represents an opportunity to collect clinical data and blood for biomarker discovery. Multiple omics technologies may be employed here including transcriptomics, proteomics, or metabolomics. We will need to see how these biomarkers perform as prospective markers of disease severity or of response to therapy.124
Conclusion
Despite recent advances that have expanded upon our understanding of IPF pathogenesis, as well as clinical trials demonstrating the first effective therapies aside from lung transplantation, we still have an incomplete understanding of this devastating disease. For this particular reason, further investigations with biomarkers will not only impact clinical care but also answer some of the most basic questions of IPF pathogenesis. This review, which has attempted to analyze the data surrounding some of the best-studied biomarkers, universally suggests that no one biomarker is yet ready for regular clinical practice. For clinicians, biomarkers that could aid in the accurate diagnosis of IPF, as well as those that could assist in early prognostication, would be of immense benefit. The review suggests that we are without strong evidence to support use of biomarkers in sorting through a differential diagnosis. On the contrary, the evidence does suggest that we are nearing development of viable clinical prediction tools, which could aid in selection of patients for treatment initiation and prioritization in transplantation. It may be possible to create a strong model using pooled results of several biomarkers that independently appear to provide clinical prognostic information.56 Finally, with the recent development and approval of IPF therapies, we have entered a new and very exciting era for IPF care where biomarker studies can begin to focus on response to therapy.
Disclosure
Dr Kass is the site investigator for clinical trials in IPF sponsored by Gilead Sciences. The other authors report no conflicts of interest in this work.
References
Raghu G, Collard HR, Egan JJ, et al. ATS/ERS/JRS/ALAT committee on idiopathic pulmonary fibrosis. an official ats/ers/jrs/alat statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. | |
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2:566–572. | |
Selman M, King TE, Pardo A, American Thoracic Society, European Respiratory Society, American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. | |
Ryu JH, Moua T, Daniels CE, et al. Idiopathic pulmonary fibrosis: evolving concepts. Mayo Clin Proc. 2014;89:1130–1142. | |
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. | |
King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. | |
Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. | |
Thomeer M, Demedts M, Behr J, et al; Idiopathic Pulmonary Fibrosis International Group, Exploring N-Acetylcysteine I Annual (IFIGENIA) Study Group. Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. 2008;31:585–591. | |
Esbriet. 2014. Available from: http://www.equinox-group.com/examples_of_clinical_innovation/esbriet.html. Accessed 14 January, 2015. | |
Loveman E, Copley VR, Colquitt JL, et al. The effectiveness and cost-effectiveness of treatments for idiopathic pulmonary fibrosis: systematic review, network meta-analysis and health economic evaluation. BMC Pharmacol Toxicol. 2014;15:63. | |
Orens JB, Estenne M, Arcasoy S, et al; Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. International guidelines for the selection of lung transplant candidates: 2006 update – a consensus report from the pulmonary scientific council of the international society for heart and lung transplantation. J Heart Lung Transplant. 2006;25:745–755. | |
Atkinson AJ, Colburn WA, DeGruttola VG, et al; Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. | |
Horimasu Y, Hattori N, Ishikawa N, et al. Different MUC1 gene polymorphisms in German and Japanese ethnicities affect serum KL-6 levels. Respir Med. 2012;106:1756–1764. | |
Kass DJ, Kaminski N. Evolving genomic approaches to idiopathic pulmonary fibrosis: moving beyond genes. Clin Transl Sci. 2011;4:372–379. | |
Ley B, Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;307:L681–L691. | |
Ohshimo S, Yokoyama A, Hattori N, Ishikawa N, Hirasawa Y, Kohno N. KL-6, a human MUC1 mucin, promotes proliferation and survival of lung fibroblasts. Biochem Biophys Res Commun. 2005;338:1845–1852. | |
Ohnishi H, Yokoyama A, Kondo K, et al. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. 2002;165:378–381. | |
Ishii H, Mukae H, Kadota J, et al. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax. 2003;58:52–57. | |
Yokoyama A, Kondo K, Nakajima M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. 2006;11:164–168. | |
Satoh H, Kurishima K, Ishikawa H, Ohtsuka M. Increased levels of KL-6 and subsequent mortality in patients with interstitial lung diseases. J Intern Med. 2006;260:429–434. | |
Ohshimo S, Ishikawa N, Horimasu Y, et al. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med. 2014;108:1031–1039. | |
Yokoyama A, Kohno N, Hamada H, et al. Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1680–1684. | |
Okuda R, Hagiwara E, Baba T, Kitamura H, Kato T, Ogura T. Safety and efficacy of pirfenidone in idiopathic pulmonary fibrosis in clinical practice. Respir Med. 2013;107:1431–1437. | |
Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc. 2007;4:252–257. | |
Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:152–160. | |
Lawson WE, Loyd JE, Degryse AL. Genetics in pulmonary fibrosis – familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci. 2011;341:439–443. | |
Lawson WE, Grant SW, Ambrosini V, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59:977–980. | |
Brasch F, Griese M, Tredano M, et al. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J. 2004;24:30–39. | |
Stevens PA, Pettenazzo A, Brasch F, et al. Nonspecific interstitial pneumonia, alveolar proteinosis, and abnormal proprotein trafficking resulting from a spontaneous mutation in the surfactant protein C gene. Pediatr Res. 2005;57:89–98. | |
Woischnik M, Sparr C, Kern S, et al. A non-BRICHOS surfactant protein c mutation disrupts epithelial cell function and intercellular signaling. BMC Cell Biol. 2010;11:88. | |
Behera D, Kaur S, Sathyanarayana G, Majumdar S. Surfactant protein-A in lung lavage fluid obtained from patients with idiopathic pulmonary fibrosis. J Assoc Physicians India. 2002;50:1409–1412. | |
McCormack FX, King TE Jr, Bucher BL, Nielsen L, Mason RJ. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1995;152:751–759. | |
McCormack FX, King TE Jr, Voelker DR, Robinson PC, Mason RJ. Idiopathic pulmonary fibrosis. Abnormalities in the bronchoalveolar lavage content of surfactant protein A. Am Rev Respir Dis. 1991;144:160–166. | |
Phelps DS, Umstead TM, Mejia M, Carrillo G, Pardo A, Selman M. Increased surfactant protein-A levels in patients with newly diagnosed idiopathic pulmonary fibrosis. Chest. 2004;125:617–625. | |
Greene KE, King TE Jr, Kuroki Y, et al. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:439–446. | |
Kinder BW, Brown KK, McCormack FX, et al. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest. 2009;135:1557–1563. | |
Barlo NP, van Moorsel CH, Ruven HJ, Zanen P, van den Bosch JM, Grutters JC. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009;26:155–161. | |
Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143:1422–1429. | |
Pardo A, Gibson K, Cisneros J, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS One. 2005;2:e251. | |
Kadota J, Mizunoe S, Mito K, et al. High plasma concentrations of osteopontin in patients with interstitial pneumonia. Respir Med. 2005;99:111–117. | |
Hanumegowda C, Farkas L, Kolb M. Angiogenesis in pulmonary fibrosis: too much or not enough? Chest. 2012;142:200–207. | |
Fehrenbach H, Kasper M, Haase M, Schuh D, Müller M. Differential immunolocalization of VEGF in rat and human adult lung, and in experimental rat lung fibrosis: light, fluorescence, and electron microscopy. Anat Rec. 1999;254:61–73. | |
Barratt SL, Blythe T, Jarrett C, et al. S137 vascular endothelial growth factor (vegf) expression in the Ipf lung – a role for anti-angiogenic isoforms? Thorax. 2014;69(Suppl):2A73. | |
Meyer KC, Cardoni A, Xiang ZZ. Vascular endothelial growth factor in bronchoalveolar lavage from normal subjects and patients with diffuse parenchymal lung disease. J Lab Clin Med. 2000;135:332–338. | |
Koyama S, Sato E, Haniuda M, Numanami H, Nagai S, Izumi T. Decreased level of vascular endothelial growth factor in bronchoalveolar lavage fluid of normal smokers and patients with pulmonary fibrosis. Am J Respir Crit Care Med. 2002;166:382–385. | |
Ando M, Miyazaki E, Ito T, et al. Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung. 2010;188:247–252. | |
Smadja DM, Nunes H, Juvin K, et al. Increase in both angiogenic and angiostatic mediators in patients with idiopathic pulmonary fibrosis. Pathol Biol (Paris). 2014;62:391–394. | |
Dancer RC, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J. 2011;38:1461–1467. | |
Zuo F, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–6297. | |
Fujishima S, Shiomi T, Yamashita S, et al. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med. 2010;134:1136–1142. | |
McKeown S, Richter AG, O’Kane C, McAuley DF, Thickett DR. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J. 2009;33:77–84. | |
Craig VJ, Polverino F, Laucho-Contreras ME, et al. Mononuclear phagocytes and airway epithelial cells: novel sources of matrix metalloproteinase-8 (MMP-8) in patients with idiopathic pulmonary fibrosis. PLoS One. 2014;9:e97485. | |
Vuorinen K, Myllärniemi M, Lammi L, et al. Elevated matrilysin levels in bronchoalveolar lavage fluid do not distinguish idiopathic pulmonary fibrosis from other interstitial lung diseases. APMIS. 2007;115:969–975. | |
Huh JW, Kim DS, Oh YM, et al. Is metalloproteinase-7 specific for idiopathic pulmonary fibrosis? Chest. 2008;133:1101–1106. | |
Rosas IO, Richards TJ, Konishi K, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS One. 2008;5:e93. | |
Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185:67–76. | |
Chien JW, Richards TJ, Gibson KF, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. 2014;43:1430–1438. | |
Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR. The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology. J Cell Commun Signal. 2009;3:275–286. | |
Ruan K, Bao S, Ouyang G. The multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci. 2009;66:2219–2230. | |
DW H. Functional role of periostin in development and wound repair: implications for connective tissue disease. J Cell Commun Signal. 2008;2:9–17. | |
Uchida M, Shiraishi H, Ohta S, et al. Periostin, a matricellular protein, plays a role in the induction of chemokines in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;46:677–686. | |
Okamoto M, Hoshino T, Kitasato Y, et al. Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur Respir J. 2011;37:1119–1127. | |
Naik PK, Bozyk PD, Bentley JK, et al; COMET Investigators. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1046–L1056. | |
Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. | |
Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. | |
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. | |
Trimble A, Gochuico BR, Markello TC, et al. Circulating fibrocytes as biomarker of prognosis in hermansky-pudlak syndrome. Am J Respir Crit Care Med. 2014;190:1395–1401. | |
García de Alba C, Buendia-Roldán I, Salgado A, et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med. 2014;191(4):427–436. | |
Fujiwara A, Kobayashi H, Masuya M, et al. Correlation between circulating fibrocytes, and activity and progression of interstitial lung diseases. Respirology. 2012;17:693–698. | |
Moeller A, Gilpin SE, Ask K, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–594. | |
Moore BB, Kolb M. Fibrocytes and progression of fibrotic lung disease. Ready for showtime? Am J Respir Crit Care Med. 2014;190:1338–1339. | |
Schutyser E, Richmond A, Van Damme J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 2005;78:14–26. | |
Prasse A, Pechkovsky DV, Toews GB, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–792. | |
Prasse A, Pechkovsky DV, Toews GB, et al. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. 2007;56:1685–1693. | |
Prasse A, Probst C, Bargagli E, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:717–723. | |
Cai M, Bonella F, He X, et al. CCL18 in serum, BAL fluid and alveolar macrophage culture supernatant in interstitial lung diseases. Respir Med. 2013;107:1444–1452. | |
Johansen JS, Christoffersen P, Møller S, et al. Serum YKL-40 is increased in patients with hepatic fibrosis. J Hepatol. 2000;32:911–920. | |
Chupp GL, Lee CG, Jarjour N, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med. 2007;357:2016–2027. | |
Malinda KM, Ponce L, Kleinman HK, Shackelton LM, Millis AJ. Gp38k, a protein synthesized by vascular smooth muscle cells, stimulates directional migration of human umbilical vein endothelial cells. Exp Cell Res. 1999;250:168–173. | |
Recklies AD, White C, Ling H. The chitinase 3-like protein human cartilage glycoprotein 39 (HC-gp39) stimulates proliferation of human connective-tissue cells and activates both extracellular signal-regulated kinase- and protein kinase B-mediated signalling pathways. Biochem J. 2002;365:119–126. | |
Furuhashi K, Suda T, Nakamura Y, et al. Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir Med. 2010;104:1204–1210. | |
Korthagen NM, van Moorsel CH, Barlo NP, et al. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med. 2011;105:106–113. | |
Korthagen NM, van Moorsel CH, Zanen P, Ruven HJ, Grutters JC. Evaluation of circulating YKL-40 levels in idiopathic interstitial pneumonias. Lung. 2014;192:975–980. | |
Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992;307:97–101. | |
Southcott AM, Jones KP, Li D, et al. Interleukin-8. Differential expression in lone fibrosing alveolitis and systemic sclerosis. Am J Respir Crit Care Med. 1995;151:1604–1612. | |
Xaubet A, Agustí C, Luburich P, et al. Interleukin-8 expression in bronchoalveolar lavage cells in the evaluation of alveolitis in idiopathic pulmonary fibrosis. Respir Med. 1998;92:338–344. | |
Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Müller-Quernheim J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med. 1998;157:762–768. | |
Tsoutsou PG, Gourgoulianis KI, Petinaki E, et al. Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respir Med. 2006;100:938–945. | |
Vasakova M, Sterclova M, Kolesar L, et al. Bronchoalveolar lavage fluid cellular characteristics, functional parameters and cytokine and chemokine levels in interstitial lung diseases. Scand J Immunol. 2009;69:268–274. | |
van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med (Berl). 1996;74:13–33. | |
Shijubo N, Imai K, Aoki S, et al. Circulating intercellular adhesion molecule-1 (ICAM-1) antigen in sera of patients with idiopathic pulmonary fibrosis. Clin Exp Immunol. 1992;89:58–62. | |
Shijubo N, Imai K, Shigehara K, et al. Soluble intercellular adhesion molecule-1 (ICAM-1) in sera and bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Clin Exp Immunol. 1994;95:156–161. | |
Tsoutsou PG, Gourgoulianis KI, Petinaki E, et al. ICAM-1, ICAM-2 and ICAM-3 in the sera of patients with idiopathic pulmonary fibrosis. Inflammation. 2004;28:359–364. | |
Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424:398–405. | |
Suzuki K, Okuno T, Yamamoto M, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature. 2007;446:680–684. | |
Czopik AK, Bynoe MS, Palm N, Raine CS, Medzhitov R. Semaphorin 7A is a negative regulator of T cell responses. Immunity. 2006;24:591–600. | |
Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. | |
Reilkoff RA, Peng H, Murray LA, et al. Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-β1-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:180–188. | |
Trujillo G, Hartigan AJ, Hogaboam CM. T regulatory cells and attenuated bleomycin-induced fibrosis in lungs of CCR7-/- mice. Fibrogenesis Tissue Repair. 2010;3:18. | |
Lo Re S, Lecocq M, Uwambayinema F, et al. Platelet-derived growth factor-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med. 2011;184:1270–1281. | |
Kotsianidis I, Nakou E, Bouchliou I, et al. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:1121–1130. | |
Garibaldi BT, D’Alessio FR, Mock JR, et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35–43. | |
Gilani SR, Vuga LJ, Lindell KO, et al. CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS One. 2010;5:e8959. | |
Herazo-Maya JD, Noth I, Duncan SR, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. | |
Hobbs JR, Turner-Warwick M. Assay of circulating immunoglobulins in patients with fibrosing alveolitis. Clin Exp Immunol. 1967;2:645–652. | |
Haslam PL, Thompson B, Mohammed I, et al. Circulating immune complexes in patients with cryptogenic fibrosing alveolitis. Clin Exp Immunol. 1979;37:381–390. | |
Kahloon RA, Xue J, Bhargava A, et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med. 2013;187:768–775. | |
Xue J, Kass DJ, Bon J, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol. 2013;191:2089–2095. | |
DePianto DJ, Chandriani S, Abbas AR, et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. 2015;70:48–56. | |
Vuga LJ, Tedrow JR, Pandit KV, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;189:966–974. | |
Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. | |
Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–1577. | |
Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–2239. | |
Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. | |
Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, et al; Pirfenidone Clinical Study Group. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–656. | |
Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:729–737. | |
Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–13056. | |
Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–317. | |
Utz JP, Ryu JH, Douglas WW, et al. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur Respir J. 2001;17:175–179. | |
Bando M, Ohno S, Hosono T, et al. Risk of acute exacerbation after video-assisted thoracoscopic lung biopsy for interstitial lung disease. J Bronchology Interv Pulmonol. 2009;16:229–235. | |
Ghatol A, Ruhl AP, Danoff SK. Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: a case series and comprehensive review of the literature. Lung. 2012;190:373–380. | |
Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res. 2011;157:191–199. | |
Li P, Li J, Chen T, et al. Expression analysis of serum microRNAs in idiopathic pulmonary fibrosis. Int J Mol Med. 2014;33:1554–1562. | |
Kass DJ, Kaminski N. Omics. In: Kolb M, Vogelmeier CF, editors. Outcomes in Clinical Trials. Sheffield: European Respiratory Monograph: European Respiratory Society; 2013:179–187. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.