")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 8
Identification of highly conserved regions in L-segment of Crimean–Congo hemorrhagic fever virus and immunoinformatic prediction about potential novel vaccine
Authors Oany AR , Ahmad SAI, Hossain MU, Jyoti TP
Received 1 October 2014
Accepted for publication 5 December 2014
Published 8 January 2015 Volume 2015:8 Pages 1—10
DOI https://doi.org/10.2147/AABC.S75250
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Juan Fernandez-Recio
Arafat Rahman Oany,1 Shah Adil Ishtiyaq Ahmad,1 Mohammad Uzzal Hossain,1 Tahmina Pervin Jyoti2
1Department of Biotechnology and Genetic Engineering, Faculty of Life Science, Mawlana Bhashani Science and Technology University, Santosh, Tangail, Bangladesh; 2Biotechnology and Genetic Engineering Discipline, Life Science School, Khulna University, Khulna, Bangladesh
Abstract: Crimean–Congo hemorrhagic fever (CCHF) is a tick-borne zoonotic viral disease with a disease fatality rate between 15% and 70%. Despite the wide range of distribution, the virus (CCHFV) is basically endemic in Africa, Asia, eastern Europe, and the Middle East. Acute febrile illness associated with petechiae, disseminated intravascular coagulation, and multiple-organ failure are the main symptoms of the disease. With all these fatal effects, CCHFV is considered a huge threat as no successful therapeutic approach is currently available for the treatment of this disease. In the present study, we have used the immunoinformatics approach to design a potential epitope-based vaccine against the RNA-dependent RNA polymerase-L of CCHFV. Both the T-cell and B-cell epitopes were assessed, and the epitope “DCSSTPPDR” was found to be the most potential one, with 100% conservancy among all the strains of CCHFV. The epitope was also found to interact with both type I and II major histocompatibility complex molecules and is considered nonallergenic as well. In vivo study of our proposed peptide is advised for novel universal vaccine production, which might be an effective path to prevent CCHF disease.
Keywords: single-stranded RNA, immunoinformatics, RNA-dependent RNA polymerase, epitope
Introduction
Crimean–Congo hemorrhagic fever virus (CCHFV) is the causative agent of the hemorrhagic fever, which was first described in Cremia in 1945.1 The virus consists of a negative-sense single-stranded RNA, and its tripartite genome comprises the small (S), medium (M), and large (L) segments, which encode the viral nucleocapsid (N), glycoprotein precursor, and polymerase-L proteins, respectively.2 Africa, Asia, eastern Europe, and the Middle East are the hotspots of this viral infection, with a high fatality rate between 15% and 70%.3,4 In case of human infection, the progression of this disease is very rapid and it causes acute febrile illness, associated with petechiae, ecchymosis, disseminated intravascular coagulation, and multiple-organ failure.5 A wide range of ticks that belong to the Hyalomma genus are considered the main vectors of this viral disease and the disease is spread through the bite of these ticks to humans or other domestic animals.6 Due to inadequate hospital care, the disease is also spread nosocomially and a case reported that, in South Africa, about 33% medical personnel are infected through needlestick injury.7 Alternate routes of infection, such as aerosol and droplet respiratory route of infection, were also suspected for several cases in Russia.8 With a broad geographic distribution, this virus already covers >30 countries across Africa, southeastern Europe, the Middle East, western Asia, and more recently, some places of southwestern Europe, particularly Spain.9–11 The wide distribution of this virus indicates the ability of the tick hosts to adapt across different environmental and geographic regions.12 Despite the outbreaks, there is no reliable vaccine or drug for the treatment of CCHFV infection in animals or humans.13 As a single-stranded-RNA-containing genome and due to the error-prone nature of its polymerase, the CCHFV incorporates random mutations into the genome. The rate of recombination in its RNA is also very high. As a consequence, the development of a vaccine or antiviral drug against CCHFV is very difficult.14
The design of epitope-based vaccines against some deadly viruses has gained much popularity due to its increased potency and safety.15,16 The application of bioinformatics in immunology, which is also termed immunoinformatics, is now widely accepted. Immunoinformatics can assist in designing new vaccines through the identification of potential T-cell epitope, B-cell epitope, and human leukocyte antigen (HLA) ligands.17–19 This novel approach has proven its efficacy in the case of multiple sclerosis,20 malaria,21 human immunodeficiency virus,22 and tuberculosis,23 with desired results. In our present study, we have proposed the design of a potential conserved epitope candidate through the immunoinformatics approaches in order to minimize the fatal effects of the CCHFV, with the expectation of finding a novel candidate for the vaccine via wet laboratory validation.
Materials and methods
Sequence retrieval and conserved region identification
UniProtKB24 database was used for the retrieval of the sequences of RNA-dependent RNA polymerase-L25 and the envelope glycoprotein26 of the CCHFV in the FASTA protein format.
BioEdit v7.2.3 sequence alignment editor27 was used for the identification of the conserved region among the sequences through multiple-sequence alignment (MSA) with ClustalW.28 Finally, Jalview v2 tool29 was used to retrieve the alignment and the CLC Sequence Viewer v7.0.2 (http://www.clcbio.com) was used for analysis of the divergence among the different strains of the CCHFV.
Antigenicity determination of the conserved peptides
VaxiJen v2. 0, a Web-based server,30 was used for the determination of the antigenicity of the conserved sequences. Herein, we used the default parameters for the prediction, with a threshold value of 0.4.
T-cell epitope prediction
Two online servers were used for the prediction of the T-cell epitope. Initially, the NetCTL v1.2 server was used for the identification of the potential T-cell epitope.31 We used the default approach to predict the epitopes, including major histocompatibility complex class I (MHC-I) binding, proteasomal C terminal cleavage, and transporter of antigenic peptide (TAP) transport efficiency. The epitope prediction was restricted to 12 MHC-I supertypes. MHC-I binding and proteasomal cleavage were performed through artificial neural networks and the weight matrix was used for estimating TAP transport efficiency. The threshold for epitope identification was set at 0.5 to maintain sensitivity and specificity of 0.89 and 0.94, respectively. Finally, CTLPred32 was implemented additionally for further confirmation about the prediction with default parameters.
MHC-I and MHC-II restriction analysis
T Cell Epitope Prediction Tools from Immune Epitope Database and Analysis Resource (IEDB-AR) was used for the prediction of MHC-I33 and MHC-II34,35 binding of the peptide. The Stabilized Matrix Method36 was used to calculate the half-maximal inhibitory concentration (IC50) of peptide binding to MHC-I molecules from different prediction methods, with a preselected 9.0-mer epitope. In case of MHC-II binding analysis, the IEDB-recommended method was used for the specific HLA-DQ, HLA-DP, and HLA-DR loci. Here, we used specific peptides for the prediction of MHC-II interaction on the basis of the antigenic conservancy and MHC-I analysis.
B-cell epitope prediction
B-cell epitope initiates immunoresponse through the interaction with B lymphocytes and causes the differentiation of B lymphocytes into plasma and memory cells.37 IEDB-AR hosts a number of Web-based tools for the prediction of B-cell epitope. Multiple tools, including the Kolaskar and Tongaonkar antigenicity scale,38 Emini surface accessibility prediction39 and Bepipred linear epitope prediction analysis, were used for the B-cell epitope prediction with high accuracy.40
Homology modeling and protein variability determination of the conserved region
Homology model of the conserved region was obtained by MODELLER v9,41 and the predicted model was assessed by PROCHECK42 and QMEAN43 servers of the SWISS-MODEL Workspace.44 For the disorder prediction among the amino acid sequences, DISOPRED v345 was used. The Protein variability server was used to calculate protein variability index using Shannon variability coefficient.46
Allergenicity and epitope conservancy analysis
In order to predict the allergenicity of the proposed epitopes with high accuracy, a Web-based server AlgPred47 was used. Herein, we used a hybrid prediction (SVMc + IgEepitope + ARPs BLAST + MAST) approach to predict the allergenicity with an accuracy of about 86% at a threshold value of −0.4. The prediction procedure follows the guidelines of the Food and Agriculture Organization/World Health Organization, 2003. A Web-based tool from IEDB-AR48 was used in order to identify the specific conservancy of the proposed epitopes.
Results
Analysis of the retrieved sequences and their divergence
A total of 80 envelope glycoproteins and 34 RNA-dependent RNA polymerase-L molecules from different variants of the CCHFV were retrieved from the UniProt database. The MSA of two different types of proteins was retrieved from BioEdit tool through ClustalW with 1,000 bootstrap replicates (Figures S1 and S2). Conserved regions were grouped for the antigenic property analysis. CLC Sequence Viewer was used to construct phylograms for both proteins from the MSA obtained from BioEdit, in order to analyze the divergence among the retrieved sequences. Phylogram of RNA-dependent RNA polymerase-L is depicted in Figure 1 and the phylogram for the envelope glycoprotein is provided in Figure S3.
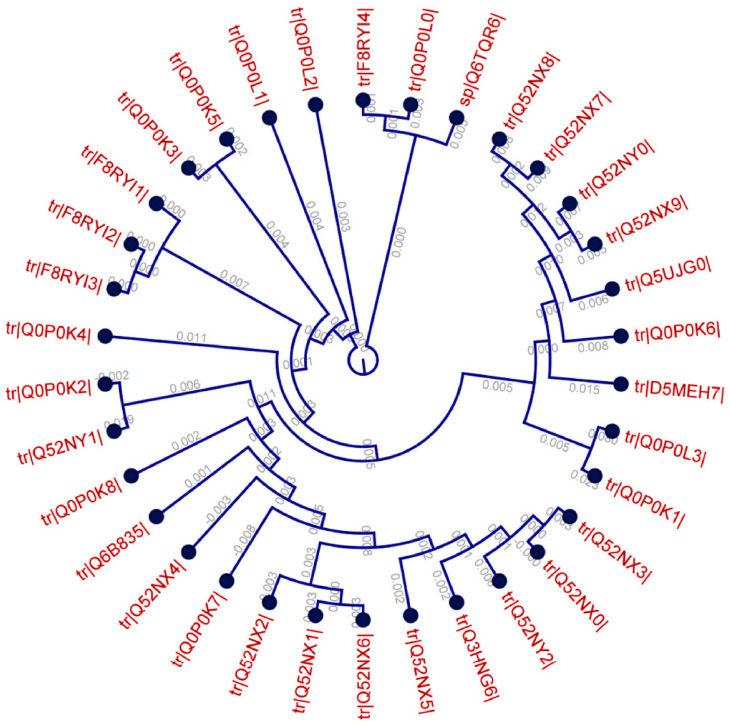 | Figure 1 Phylogenetic tree showing the evolutionary divergence among the different RNA-dependent RNA polymerase-L molecules of the CCHFV. |
Antigenic peptide identification
Initially, the conserved sequences (MSA number: 3,563–3,915) of RNA-dependent RNA polymerase-L were separated into four conserved peptides, according to their continuity in the MSA. Then the VaxiJen v2.0 server was used to predict the antigenicity of all the grouped conserved peptides (Table 1). On the basis of the VaxiJen score, the top two conserved peptides (MSA number: 3,563–3,658 and 3,694–3,773) were selected for further analysis. The peptide with second-best VaxiJen score (MSA: 3,694–3,773) showed better results during further analysis of T-cell epitope identification, MHC interaction analysis, and B-cell epitope identification. The conservancy of this region among the different viral strains is partially depicted in Figure 2.
 | Table 1 Antigenicity determination of the conserved peptide by Vaxijen server |
 | Figure 2 MSA of the conserved region of RNA-dependent RNA polymerase-L. Only the partial sequences containing the proposed epitope sequence are shown here. |
T-cell epitope identification and MHC interaction analysis
NetCTL v1.2 server predicted the T-cell epitopes through the combined approach for the 12 MHC-I supertypes. On the basis of the combined score, five epitopes with top scores (Table 2) were selected for further analysis. CTLPred server also predicted the T-cell epitopes based on an approach that combined artificial neural networks and support vector machines (Table 3). From the analysis, the common epitope–containing peptide, which was predicted by both servers, was selected and used for the MHC-binding analysis.
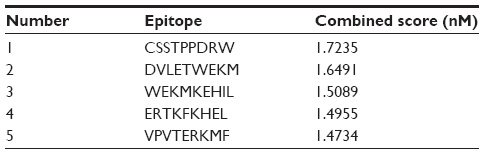 | Table 2 Prediction of the T-cell epitope by NetCTL server on the basis of combined score |
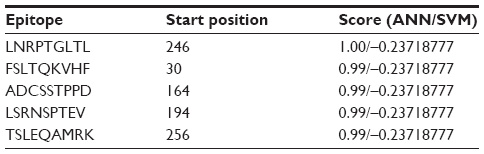 | Table 3 Prediction of the T-cell epitope by CTLPred server |
MHC-I-binding prediction, which was run through the Stabilized Matrix Method, predicted a wide range of MHC-I allele interactions for the proposed T-cell epitopes. The MHC-I alleles for which the epitope showed higher affinity (IC50 <200 nM) are listed in Table 4. The output of the MHC-II interaction analysis is also shown in Table 4.
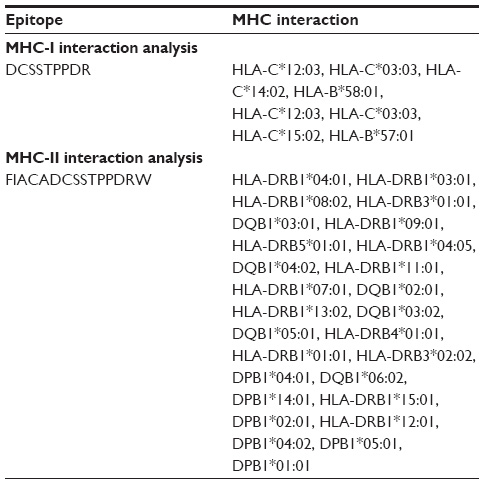 | Table 4 MHC-I and MHC-II interaction of the proposed sequence by IEDB-AR |
B-cell epitope identification
Amino acid–based methods were used for the prediction of potential B-cell epitope. Antigenic property of the peptides was assessed using the Kolaskar and Tongaonkar antigenicity scale. The average antigenic propensity score of the protein was 1.019, with a maximum of 1.197 and a minimum of 0.884. The threshold value for antigenic determination of the peptide was 1.0, where all epitopes with values >1.0 were potential antigenic determinants. We found that 14 epitopes satisfied the threshold value set prior to the analysis, and they had the potential to express the B-cell response. The results are summarized in Table 5 and Figure 3. Peptides with the potential to function as B-cell epitope must be surface accessible. For this reason, Emini surface accessibility prediction was employed, with a maximum propensity score of 4.946 at threshold 1.0. Results are summarized in Figure 4. Finally, Bepipred linear epitope prediction tool predicted the linear B-cell epitope with the most reliable results of prediction. Results are shown in Figure 5.
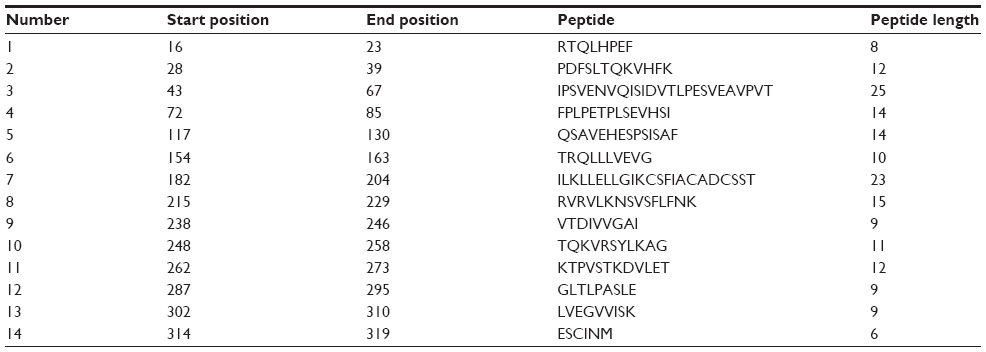 | Table 5 Kolaskar and Tongaonkar antigenicity analysis |
 | Figure 3 Kolaskar and Tongaonkar antigenicity prediction of the conserved peptide, ranging from 3,563 to 3,915 MSA number. |
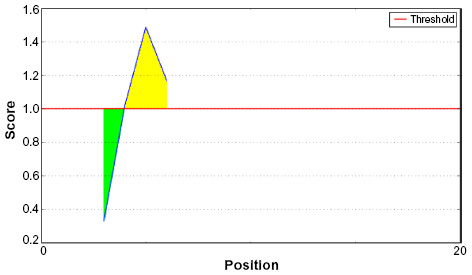 | Figure 4 Emini surface accessibility prediction of the proposed epitope, with a minimum propensity score of 0.327 and maximum score of 1.488. |
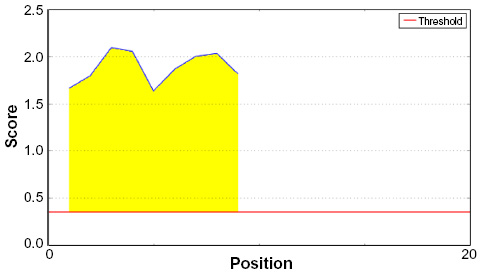 | Figure 5 Bepipred linear epitope prediction of the proposed epitope with a minimum propensity score of 1.631 and maximum score of 2.094. |
Structure analysis and protein variability determination
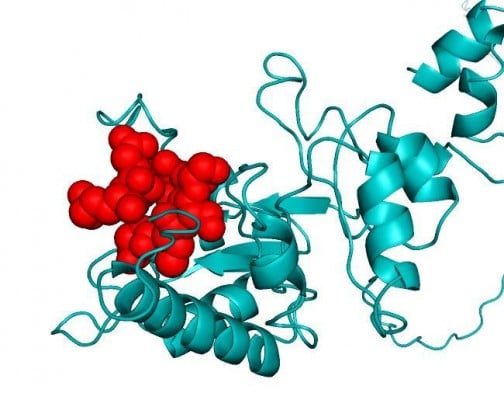
Homology model of the conserved region was obtained by the MODELLER software, which is shown in Figure 6A. PROCHECK server validated the stereochemical quality of the model through Ramachandran Plot (Figure 6B), and Qmean server also assessed the tertiary structure, with a Qmean6 score of 0.268. DISOPRED v3 server predicted the disorder of the conserved peptide in order to get insight about the disorder among the conserved sequences, which is depicted in Figure 7. Protein variability server predicted the variability of the conserved region of the RNA-dependent RNA polymerase-L (Figure 8) to ensure that the proposed epitope is within the invariable region.
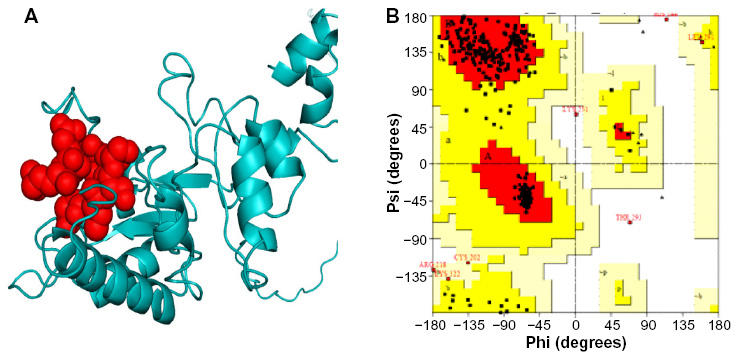 | Figure 6 Three-dimensional structure prediction and validation. |
 | Figure 7 Disorder prediction of the conserved antigenic amino acid sequences. Here, our proposed epitope lies outside (197–202) of the disordered region to secure its potentiality as an effective epitope. |
 | Figure 8 Protein variability index of the conserved peptides of all the sequences. The prediction suggests that our proposed epitope (197–202) falls in the invariable region. |
Epitope conservancy and allergenicity analysis
IEDB conservancy analysis tool analyzed the epitope conservancy of the proposed epitopes that are shown in Table 6. AlgPred predicted the allergenicity of the epitopes based on amino acid composition. The prediction score of AlgPred for the two epitopes in combination was 0.49752311 at threshold of –0.4.
 | Table 6 Epitope conservancy analysis |
Discussion
With a widely distributed endemically affected region and a randomly mutated genome, CCHFV imposes a great challenge to researchers in developing a successful therapeutic approach against it. The ability of an epitope-based vaccine to stimulate an effective specific immune response with a minute structure and without any unexpected side effects has made it a good choice for vaccine development.49 In this instance, we started with the preferable target, namely, the envelope glycoprotein, but failed to identify any unique conserved region (Figure S1; multiple-sequence alignment of the envelope glycoprotein of CCHFV.) to design a peptide vaccine against the envelope glycoprotein. This was also revealed by phylogeny analysis, which is shown in Figure S3; phylogenetic tree showing the evolutionary divergence among the different envelope glycoproteins of CCHFV. RNA-dependent RNA polymerase, a product of the L-segment of the genome, comprises a unique conserved region among all the available strains of CCHFV (Figure S2; multiple-sequence alignment of the RNA-dependent RNA polymerase-L of CCHFV). This was the pedestal to think about a novel vaccine candidate. To ensure a firm immune response, we looked for the activation of both T-cell and B-cell immunity with a single epitope.50 Antigenicity of the conserved peptides indicated their ability to provoke potential immune response and they were used for further analysis involving T-cell epitope prediction. Through the analysis of the output of both NetCTL and CTLPred, it was found that the epitope “DCSSTPPDR” would be the best candidate for the activation of T-cell immunity with potential antigenicity. Analysis of the MHC ligands for both type I and II revealed that the core epitope “DCSSTPPDR” would interact with the highest number of HLA molecules and that it would support the MHC molecules to present the epitope on the T-cell surface. The complete peptide for MHC-II restriction was FIACADCSSTPPDRW. The peptide DCSSTPPDR was also found to be the most potential candidate to raise B-cell immune response by amino acid–based B-cell epitope prediction, including Kolaskar and Tongaonkar antigenicity scale, Emini surface accessibility prediction, and Bepipred linear epitope prediction.
In order to see the exact location of the proposed epitope of the protein (shown spherically in Figure 6A), the three-dimensional structure of the conserved peptide was modeled. This predicted model was validated with Ramachandran Plot (Figure 6B), whereby 89.8% amino acid residues were found within the favored region. The disorderliness of the peptide remains outside of the proposed epitope region, which would secure the functioning of our predicted epitope (Figure 7).
The most important feature of an epitope enabling its use as a vaccine is its conservancy. Conservancy analysis of the proposed epitope found 100% conservancy among all the available sequences. Another important criterion of the peptide vaccine is its allergenicity51; our proposed epitope was examined in silico and found to be nonallergenic in nature. A recent study in designing a vaccine against CCHFV targeting the envelope glycoprotein showed a high success rate in a mouse model.52 But because it is a structural protein, the rate of mutation is higher than that of nonstructural proteins such as RNA polymerase,53 a phenomenon that has been evidenced in our study through sequence analysis. As our proposed epitope is shown to be 100% conserved among different CCHF strains, we suggest that it will be the best possible candidate for vaccine designing.
Epitope-based vaccine designing is now becoming more popular and already has been established for rhinovirus,54 dengue virus,55 human corona virus,56 and some others. This type of work has also been proven in vitro.57 So, considering the above analysis,we predict that our proposed epitope would also trigger an immune response in vitro.
Conclusion
The findings from this study denote that integrated computational approaches are very much effective for designing vaccine candidates against some deadly viruses such as the CCHFV, with the formally delineated experimental procedure. Thus, co mputational studies save both time and cost for researchers and can lead the wet laboratory work with higher possibilities of getting the desired outcome.
Disclosure
The authors report no conflicts of interests in this work.
References
Cunha BA. Tickborne Infectious Diseases: Diagnosis and Management. Vol 24. Boca Raton, FL: CRC Press; 2000. | |
Schmaljohn C, Hooper J. Bunyaviridae: the viruses and their replication. Fields Virol. 2001;2(2):20. | |
Lacy MD, Smego R. Viral hemorrhagic fevers. Adv Pediatr Infect Dis. 1995;12:21–53. | |
Centers for Disease Control and Prevention (CDC). Management of patients with suspected viral hemorrhagic fever. MMWR Morb Mortal Wkly Rep. 1988;37:1. | |
Ergönül Ö. Crimean–Congo haemorrhagic fever. Lancet Infect Dis. 2006;6(4):203–214. | |
Ergonul O. Crimean–Congo hemorrhagic fever virus: new outbreaks, new discoveries. Curr Opin Virol. 2012;2(2):215–220. | |
Swanepoel R, Struthers J, Shepherd A, McGillivray G, Nel M, Jupp P. Crimean–Congo hemorrhagic fever in South Africa. Am J Trop Med Hyg. 1983;32(6):1407–1415. | |
Whitehouse CA. Crimean–Congo hemorrhagic fever. Antiviral Res. 2004;64(3):145–160. | |
David-West TS, Cooke AR, David-West AS. Seroepidemiology of Congo virus (related to the virus of Crimean haemorrhagic fever) in Nigeria. Bull World Health Organ. 1974;51(5):543. | |
Gonzalez J-P, LeGuenno B, Guillaud M, Wilson ML. A fatal case of Crimean–Congo haemorrhagic fever in Mauritania: virological and serological evidence suggesting epidemic transmission. Trans R Soc Trop Med Hyg. 1990;84(4):573–576. | |
Wilson ML, LeGuenno B, Guillaud M, Desoutter D, Gonzalez J-P, Camicas J-L. Distribution of Crimean–Congo hemorrhagic fever viral antibody in Senegal: environmental and vectorial correlates. Am J Trop Med Hyg. 1990;43(5):557–566. | |
Estrada-Peña A, Ruiz-Fons F, Acevedo P, Gortazar C, la Fuente J. Factors driving the circulation and possible expansion of Crimean–Congo haemorrhagic fever virus in the western Palearctic. J Appl Microbiol. 2013;114(1):278–286. | |
Hoogstraal H. The epidemiology of tick-borne Crimean–Congo hemorrhagic fever in Asia, Europe, and Africa. J Med Entomol. 1979;15(4):307–417. | |
Kraus AA, Mirazimi A. Molecular biology and pathogenesis of Crimean–Congo hemorrhagic fever virus. Future Virol. 2010;5(4):469–479. | |
Holland J, Domingo E. Origin and evolution of viruses. Virus Genes. 1998;16(1):13–21. | |
Sette A, Newman M, Livingston B, et al. Optimizing vaccine design for cellular processing, MHC binding and TCR recognition. Tissue Antigens. 2002;59(6):443–451. | |
Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15(4):461–470. | |
Poland GA, Ovsyannikova IG, Jacobson RM. Application of pharmacogenomics to vaccines. Pharmacogenomics. 2009;10(5):837–852. | |
Petrovsky N, Brusic V. Computational immunology: the coming of age. Immunol Cell Biol. 2002;80(3):248–254. | |
Bourdette DN, Edmonds E, Smith C, et al. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler. 2005;11(5):552–561. | |
López JA, Weilenman C, Audran R, et al. A synthetic malaria vaccine elicits a potent CD8+ and CD4+ T lymphocyte immune response in humans. Implications for vaccination strategies. Eur J Immunol. 2001;31(7):1989–1998. | |
Wilson CC, McKinney D, Anders M, et al. Development of a DNA vaccine designed to induce cytotoxic T lymphocyte responses to multiple conserved epitopes in HIV-1. J Immunol. 2003;171(10):5611–5623. | |
Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med. 2005;11:S25–S32. | |
Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(suppl 1):D115–D119. | |
Kinsella E, Martin SG, Grolla A, Czub M, Feldmann H, Flick R. Sequence determination of the Crimean–Congo hemorrhagic fever virus L segment. Virology. 2004;321(1):23–28. | |
Sanchez AJ, Vincent MJ, Nichol ST. Characterization of the glycoproteins of Crimean–Congo hemorrhagic fever virus. J Virol. 2002;76(14):7263–7275. | |
Hall TA. BioEdit: A User-Friendly Biological Sequence Alignment Editor and analysis Program for Windows 95/98/NT. Paper presented at: Nucleic Acids Symposium Series; 1999; London. | |
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. | |
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. | |
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):4. | |
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8(1):424. | |
Bhasin M, Raghava G. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22(23):3195–3204. | |
Buus S, Lauemøller SL, Worning P, et al. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62(5):378–384. | |
Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. 2010;11(1):568. | |
Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048. | |
Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6(1):132. | |
Nair DT, Singh K, Siddiqui Z, Nayak BP, Rao KV, Salunke DM. Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J Immunol. 2002;168(5):2371–2382. | |
Kolaskar A, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1):172–174. | |
Emini EA, Hughes JV, Perlow D, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55(3):836–839. | |
Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2(1):2. | |
Šali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23(3):318–326. | |
Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8(4):477–486. | |
Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343–350. | |
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. | |
Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–2139. | |
Garcia-Boronat M, Diez-Rivero CM, Reinherz EL, Reche PA. PVS: a web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Res. 2008;36(Suppl 2):W35–W41. | |
Saha S, Raghava G. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34(Suppl 2):W202–W209. | |
Bui H-H, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007; 8(1):361. | |
Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78(15):8312–8321. | |
Arnon R. A novel approach to vaccine design – epitope-based vaccines. FEBS J. 2006;273:33–34. | |
McKeever TM, Lewis SA, Smith C, Hubbard R. Vaccination and allergic disease: a birth cohort study. Am J Public Health. 2004; 94(6):985. | |
Buttigieg KR, Dowall SD, Findlay-Wilson S, et al. A novel vaccine against Crimean–Congo haemorrhagic fever protects 100% of animals against lethal challenge in a mouse model. PLoS One. 2014;9(3):e91516. | |
Lu R, Yu X, Wang W, et al. Characterization of human coronavirus etiology in Chinese adults with acute upper respiratory tract infection by real-time RT-PCR assays. PLoS One. 2012;7(6):e38638. | |
Lapelosa M, Gallicchio E, Arnold GF, Arnold E, Levy RM. In silico vaccine design based on molecular simulations of rhinovirus chimeras presenting HIV-1 gp41 epitopes. J Mol Biol. 2009;385(2):675–691. | |
Chakraborty S, Chakravorty R, Ahmed M, et al. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 2010;10(5):235–246. | |
Oany AR, Emran AA, Jyoti TP. Design of an epitope-based peptide vaccine against spike protein of human corona virus: an in silico approach. Drug Des Devel Ther. 2014;8:1139–1149. | |
Khan MK, Zaman S, Chakraborty S, et al. In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol Immunol. 2014;61(1):16–22. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.