")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Granulomatosis with polyangiitis: rapidly progressive necrotizing glomerulonephritis in a pediatric patient
Authors Luna M, Bocanegra V, Vallés P
Received 6 November 2013
Accepted for publication 2 December 2013
Published 23 April 2014 Volume 2014:7 Pages 153—156
DOI https://doi.org/10.2147/IJNRD.S57109
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mariana Luna,1 Victoria Bocanegra,3 Patricia G Vallés1,2
1Nephrology Division, Pediatric Department, Dr Humberto Notti Pediatric Hospital, Mendoza, Argentina; 2Pathophysiology Area, Pathology Department, School of Medicine, National Cuyo University, Mendoza, Argentina; 3National Council of Scientific and Technical Research of Argentina (CONICET), Buenos Aires, Argentina
Abstract: Granulomatosis with polyangiitis (GPA) is associated with a broad range of clinical manifestations including renal disease. It is a systemic vasculitis that is rarely encountered in children. We present a 14-year-old girl who suffered from pharyngitis 1 week before admittance to hospital. She was admitted for macroscopic hematuria and oliguria, under the possibility of nephritic syndrome. Renal failure with rapidly progressive glomerulonephritis occurred within 24 hours. Immunologic tests showed the presence of type-C anti-neutrophil cytoplasmic antibodies (c-ANCA with antiproteinase 3 specificity) and renal biopsy revealed pauci-immune crescentic focal necrotizing glomerulonephritis. Treatment including methylprednisolone and cyclophosphamide intravenous pulses allowed renal recovery after 3 weeks. The clinical, hematological, and biochemical parameters improved substantially, achieving remission. Granulomatosis with polyangiitis, although rare in children, should be considered in the above clinical scenario. This case underlines that knowledge of renal histology diagnosis and early aggressive immunosuppressive therapy are essential for the management of these patients.
Keywords: acute renal failure, vasculitis, crescentic pauci-immune glomerulonephritis-Type-C, antineutrophil cytoplasmic antibodies (c-ANCA)-macroscopic hematuria
Introduction
Granulomatosis with polyangiitis (GPA), formerly called Wegener’s granulomatosis, is a rare autoimmune disorder of unknown etiology that is characterized by granulomatous inflammation and antineutrophil cytoplasmatic antibodies (ANCA) associated with small vessel vasculitis. ANCA are mainly directed against proteinase 3 (PR3). GPA has a broad clinical spectrum that ranges from predominantly granulomatous manifestations restricted to the respiratory tract, to severe, life-threatening necrotizing vasculitis affecting many organs, with a predilection for lung and kidney involvement. The disease mostly affects adults aged 25–50 years and is rare in children.1
The European classification criteria for granulomatosis with polyangiitis requires the diagnosis of three of the following six features: 1) abnormal urinalysis; 2) granulomatous inflammation on biopsy; 3) nasal sinus inflammation; 4) subglottic, tracheal, or endobronchial stenosis; 5) abnormal chest imaging; and 6) positive ANCA testing.2
Case report
The patient is a 14-year-old girl diagnosed with pharyngitis 1 week before admission. She was referred to the Dr Humberto Notti Pediatric Hospital for macroscopic hematuria accompanied by oliguria, with suspicion of nephritic syndrome. On examination, the patient appeared healthy with a body mass index of 22.5 kg/m2, with no fever or edema. Her blood pressure was 110/90 mmHg (normal blood pressure levels for age, height and sex), with dispersed abdominal pain, aching shoulders, and myalgias. Family history was not significant and there was no history of illicit drugs.
Initial laboratory data were as follows: serum creatinine 3.10 mg/dL, urea 50 mg/dL, hemoglobin concentration 10.3 g/dL, and urinalysis with more than 100 red blood cells, some of them dysmorphic without crystals or hyaline cylinders. Negative 24-hour proteinuria was reported. Laboratory data 12 hours after admission into hospital were as follows: serum creatinine 5.7 mg/dL, urea 85 mg/dL, leukocytes 12,500/μL, platelet count 115 × 103/μL, C-reactive protein (CRP) 2.27 mg/dL (227.3 mg/L), ferritin 313 ng/mL (6–70 ng/mL), fibrinogen 476 mg% (200–400 mg%), potassium 3.4 mEq/L, sodium 133 mEq/L, chloride 93 mEq/L, calcium 8.59 mg/dL, acid base balance pH 7.4/PCO2 29.7 mmHg/HCO3 20 mmol/L, lactate dehydrogenase 800 U/L, and decreased glomerular filtration rate 14.4 mL/m/1.73 m2 by Schwartz formula. Chest radiograph with diffuse interstitial infiltrate in both lungs was demonstrated. Renal ultrasound showed increased kidney size with increased renal parenchymal echogenicity consistent with parenchymal disease.
Twenty-four hours after admission, peritoneal dialysis was started. The next day, creatinine and uremia continued to increase. A percutaneous renal biopsy was performed and intravenous steroid pulses at 10 mg/kg/day were begun.
Serology tests demonstrated the presence of positive C-ANCA by indirect immunofluorescence with documented proteinase 3–specific ANCA (PR3-ANCA). P-ANCA and peroxidase-specific antineutrophil cytoplasmic autoantibodies (MPO-ANCA) were negative. Rheumatoid factor and C3, C4 complement levels were normal. Anti-DNA antibody titers measured by enzyme-linked immunosorbent assay (ELISA) and anti-nuclear antibody (ANA) by immunofluorescence were at normal levels. Viral serology (hepatitis B and C, HIV), and blood and throat cultures were negative. Anti-streptolysin O (ASO) titers were within the normal limits.
Diagnosis of renal involvement was confirmed through biopsy. Microscopic optic study in 20 glomeruli revealed renal parenchyma with less than 50% glomeruli presenting segmental capillary fibrinoid necrosis with neutrophil exudate surrounding glomeruli. Tubule interstitial infiltrate was also shown (Figure 1). Cellular crescent was seen in 20% of the glomeruli (Figure 2). Immunofluorescence with antibodies against IgG, IgA, IgM, and C3 was negative for immunoglobulins, and the intensity of immunofluorescence staining was very weak (trace or trace to 1+) for C3. The final diagnosis was focal segmental necrotizing glomerulonephritis.
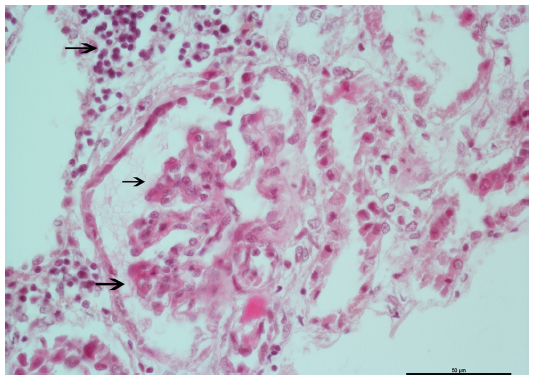 | Figure 1 Renal biopsy specimen showing focal necrotizing glomerulonephritis in the patient. |
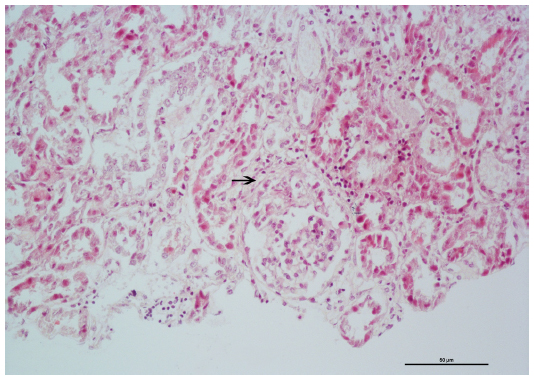 | Figure 2 Renal biopsy specimen showing mild focal segmental extracapillary proliferation (arrow); hematoxylin and eosin stain. |
To investigate other organs affected with small vessel vasculitis, nasal cavity and sinuses computer tomography (CT) showed rhinosinusitis signals and inflammatory tissue. Chest CT showed bilateral pleural effusion.
Immunosuppressive therapy was started with three methylprednisolone pulses, following by cyclophosphamide intravenous pulses (each with a dosage of 750 mg/m2 body surface) continued at monthly intervals for 12 months. After 23 days, the patient’s renal function recovered. The early normalization of the glomerular filtration rate made plasma exchange in the protocol treatment unnecessary.
Further, the remission treatment consisted of a daily dose of mycophenolate mofetil 750 mg/m2 in two divided doses and oral prednisolone (1 mg/kg for 6 weeks) followed by slow tapering. She was discharged after 5 weeks. Neither symptoms nor signs of vasculitis were seen at the last clinical control. Six months after diagnosis, the patient was negative for PR3-ANCA.
Discussion
The present report describes the development of clinical rapidly progressive glomerulonephritis and histological crescentic necrotizing glomerulonephritis in a girl. Crescentic glomerulonephritis is classified into three main categories on the basis of direct immunofluorescence microscopy. Anti-glomerular basement membrane (GBM) crescentic glomerulonephritis is characterized by the presence of circulating antibodies to GBM and linear deposition of IgG along the GBM; immune complex-mediated crescentic glomerulonephritis is characterized by the deposition of immune complexes in the glomeruli; while pauci-immune crescentic glomerulonephritis presents as focal necrotizing glomerulonephritis with little or no glomerular staining for immunoglobulins.3
Rapid progressive renal failure was an initial clinical diagnosis in this patient who suffered from progressive renal impairment in a short period of time. Although immune complex-mediated crescentic glomerulonephritis is the most common cause of rapidly progressive glomerulonephritis in children, we unusually found renal histopathology of pauci-immune crescentic glomerulonephritis in this patient. These findings are also in contrast to previous case series in children showing that immune complex glomerulonephritis constitutes the majority of cases with crescentic glomerulonephritis.4
Importantly, renal biopsy with indirect immunofluorescent staining in the patient allowed the exclusion of anti-GBM antibody disease in the setting of a pulmonary renal syndrome. Approximately three-fourths of patients with pauci-immune or ANCA-associated crescentic glomerulonephritis have systemic small-vessel vasculitis. Only ANCAs directed against PR3 or MPO have been associated with primary vasculitic syndromes. C-ANCA directed against PR3 is most specific for GPA as we have demonstrated in this patient.
However, pulmonary involvement occurred in half of the patients and subglottic stenosis is frequently present in pediatric patients at onset; these were not observed in our patient. Findings on chest radiography are abnormal in two-thirds of adults with GPA. Findings on thin-section sinus CT scans are abnormal in more than 90% of adults with GPA.
A similar number would be expected in the pediatric population, based on an 83% incidence of sinusitis.5
The clinical features identified in our patient as hematuria, slightly raised blood pressure, and the rapid loss of renal function suggest glomerulonephritis. In addition, with positive serum c-ANCA levels and kidney biopsy showing granulomatous inflammation, diagnosis of GPA can be confirmed.
In consideration of the pathogenesis, ANCA has been examined extensively ever since the soluble lysosomal enzymes myeloperoxidase and proteinase 3 were identified as targets. Antibodies to these enzymes activate primed neutrophils and cause neutrophil-dependent endothelial injury in vitro. However, the pathogenic potential of ANCA against proteinase 3, which is mainly found in patients with GPA remains unproven. Additionally, a subgroup of patients with pauci-immune crescentic glomerulonephritis persistently lack cANCA.6
Experimental studies have shown that proteinase-3-directed autoimmunity involves the complementary peptide of proteinase 3, which is encoded by the antisense strand of the proteinase 3 gene. Exposure of the immune system to this peptide triggers the formation of antibodies that cross-react with proteinase 3. DNA sequences complementary to the proteinase 3 gene have been identified in microorganisms, including Staphylococcus aureus, which supports the role of infectious agents as triggers of proteinase 3 autoimmunity via molecular mimicry.7,8 Recently, Kain et al reported that infection by fimbriated bacteria can trigger cross-reactive autoimmunity to a previously characterized ANCA antigen, lysosomal membrane protein 2 (LAMP-2), which results in the production of autoantibodies that activate neutrophils and damage human microvascular endothelium in vitro and in vivo, causing pauci-immune focal necrotizing glomerulonephritis,9 even though no infection was demonstrated by culture studies in our patient.
Our patient was treated with methylprednisolone and cyclophosphamide, which stabilized renal function after 3 weeks. Plasma exchange was not included in the protocol.
The optimal prescription of plasmapheresis for the treatment of GPA is uncertain. Clinical studies have focused on the use of plasma exchange to rescue organ function in rapidly progressive glomerulonephritis and lung hemorrhage, based on the rationale of removing circulating ANCAs.10 However, evidence derived from its use in children is scarce.11,12
Even in light of advances in the understanding of pathophysiology and serologic testing, renal biopsy remains the mainstay of the diagnosis of crescentic glomerulonephritis, and early definitive diagnosis of rapid progressive renal failure is essential to reverse the progression to end-stage kidney disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Akikusa JD, Schneider R, Harvey EA, et al. Clinical features and outcome of pediatric Wegener’s granulomatosis. Arthritis Rheum. 2007;57:837–844. | |
Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65(7):936–941. | |
Jennette J, Cohen J, Harringtom J, Madias N, Zusman C. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164–1177. | |
Dewan D, Gulati S, Sharma RK, et al. Clinical spectrum and outcome of crescentic glomerulonephritis in children in developing countries. Pediatr Nephrol. 2008;23(3):389–394. | |
Holle J, Laudien M, Gross W. Clinical manifestations and treatment of Wegener’s Granulomatosis. Rheum Dis Clin North Am. 2010;36:507–526. | |
Chen M, Kallenberg C, Zhao M. ANCA negative pauci-immune crescentic glomerulonephritis. Nat Rev Nephrol. 2009;5:313–318. | |
Bosch X, Mirapeix E. LAMP-2 illuminates pathogenesis of ANCA glomerulonephritis. Nat Rev Nephrol. 2009;5:247–249. | |
Pendergraft WF 3rd, Preston GA, Shah RR, et al. Autoimmunity is triggered by cPR-3105–3201, a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–79. | |
Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauciimmune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–1096. | |
Lapraik C, Watts R, Bacon P, et al. BSR and BHPR guidelines for the management of adults with ANCA associated vasculitis. Rheumatology (Oxford). 2007;46(10):1615–1616. | |
Wright E, Dillon MJ, Tullus K. Childhood vasculitis and plasma exchange. Eur J Pediatr. 2007;166(2):145–151. | |
Siomou E, Tramma D, Bowen C, Milford DV. ANCA-associated glomerulonephritis/systemic vasculitis in childhood: clinical features-outcome. Pediatr Nephrol. 2012;27(10):1911–2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.