")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
EGFR-targeted plasmonic magnetic nanoparticles suppress lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage
Authors Kuroda S, Tam J, Roth J, Sokolov K, Ramesh R
Received 11 April 2014
Accepted for publication 8 May 2014
Published 8 August 2014 Volume 2014:9(1) Pages 3825—3839
DOI https://doi.org/10.2147/IJN.S65990
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Shinji Kuroda,1 Justina Tam,2 Jack A Roth,1 Konstantin Sokolov,2 Rajagopal Ramesh3–5
1Department of Thoracic and Cardiovascular Surgery, 2Department of Imaging Physics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 3Department of Pathology, 4Graduate Program in Biomedical Sciences, 5Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Background: We have previously demonstrated the epidermal growth factor receptor (EGFR)-targeted hybrid plasmonic magnetic nanoparticles (225-NP) produce a therapeutic effect in human lung cancer cell lines in vitro. In the present study, we investigated the molecular mechanism of 225-NP-mediated antitumor activity both in vitro and in vivo using the EGFR-mutant HCC827 cell line.
Methods: The growth inhibitory effect of 225-NP on lung tumor cells was determined by cell viability and cell-cycle analysis. Protein expression related to autophagy, apoptosis, and DNA-damage were determined by Western blotting and immunofluorescence. An in vivo efficacy study was conducted using a human lung tumor xenograft mouse model.
Results: The 225-NP treatment markedly reduced tumor cell viability at 72 hours compared with the cell viability in control treatment groups. Cell-cycle analysis showed the percentage of cells in the G2/M phase was reduced when treated with 225-NP, with a concomitant increase in the number of cells in Sub-G1 phase, indicative of cell death. Western blotting showed LC3B and PARP cleavage, indicating 225-NP-treatment activated both autophagy- and apoptosis-mediated cell death. The 225-NP strongly induced γH2AX and phosphorylated histone H3, markers indicative of DNA damage and mitosis, respectively. Additionally, significant γH2AX foci formation was observed in 225-NP-treated cells compared with control treatment groups, suggesting 225-NP induced cell death by triggering DNA damage. The 225-NP-mediated DNA damage involved abrogation of the G2/M checkpoint by inhibiting BRCA1, Chk1, and phospho-Cdc2/CDK1 protein expression. In vivo therapy studies showed 225-NP treatment reduced EGFR phosphorylation, increased γH2AX foci, and induced tumor cell apoptosis, resulting in suppression of tumor growth.
Conclusion: The 225-NP treatment induces DNA damage and abrogates G2/M phase of the cell cycle, leading to cellular apoptosis and suppression of lung tumor growth both in vitro and in vivo. Our findings provide a rationale for combining 225-NP with other DNA-damaging agents for achieving enhanced anticancer activity.
Keywords: lung cancer, epidermal growth factor receptor, autophagy
Introduction
Application of nanotechnology to cancer medicine has progressed rapidly in recent years, resulting in testing of novel imaging contrast agents and drug delivery systems.1–3 One among several nanomaterials that has been tested and has emerged as an excellent optical contrast agent is gold-based nanoparticles (NPs).4 Gold NPs have the inherent ability to resonantly scatter visible and near infrared light and thus have been used as an optical contrast agent. Gold NPs, in addition to being used as a contrast agent, have also been tested as therapeutic agents.5 Paramagnetic iron oxide NPs are another type of NP that have been investigated for their effectiveness as an imaging agent and has been shown to produce greater contrast than conventional contrast agents such as gadolinium by magnetic resonance imaging.6 Additionally, iron oxide-based NPs have also been tested as a therapeutic agent.7 All of these studies establish the utility of iron oxide- and gold-based NPs in cancer cell imaging and therapy.
Epidermal growth factor receptor (EGFR), which is a member of the ErbB receptor tyrosine kinases, is overexpressed in approximately 50%–80% of non-small cell lung cancer and is a target for cancer therapy.8,9 Small-molecule inhibitors such as gefitinib and erlotinib, both of which are EGFR-tyrosine kinase inhibitors, have shown clinical activity.10–12 Similarly, monoclonal antibodies such as cetuximab targeted to the extracellular domain of EGFR have demonstrated clinical efficacy.13 The usefulness of these small molecule inhibitors is limited by the development of acquired resistance to therapy.14,15 Therefore, novel and improved forms of therapy are warranted.
We have previously reported the antitumor activity of EGFR-targeted hybrid plasmonic magnetic NPs (225-NP) against human lung cancer cells in vitro.16 The NPs consisted of a paramagnetic iron core that is covered by a gold layer and is functionalized with a therapeutic monoclonal anti-EGFR antibody (225). In the same study, we observed that the antitumor activity of 225-NP is greatly enhanced only when 225-antibody (225-Ab) is attached to the NP as a simple mixture of free antibody, and NP alone does not produce cytotoxic effects comparable to 225-NP. However, in that study, the molecular mechanism(s) that contributed to the 225-NP-mediated enhanced tumor cell killing was not investigated. Furthermore, the in vivo efficacy of 225-NP was not tested.
In the present study, we investigated the molecular mechanism for 225-NP-mediated cytotoxicity in vitro and the in vivo efficacy of 225-NP using a lung tumor xenograft model. We demonstrate 225-NP induced DNA damage and abrogated the G2/M phase of the cell cycle in lung cancer cells, resulting in autophagy and apoptosis in vitro. However, 225-NP, unlike conventional DNA-damaging agents such as ionizing radiation (IR) and chemotherapy, inhibited the BRCA1-Chk1 signaling, thereby overriding the G2 to M checkpoint arrest and not allowing the cells to repair the DNA damage. The resulting outcome is that the cells bearing DNA damage proceeded to the M phase as evidenced by the expression of the mitotic marker, histone H3, resulting in cellular apoptosis. In vivo studies showed 225-NP treatment induced DNA damage and apoptosis, resulting in marked suppression of tumor growth. To our knowledge, our study demonstrates the antitumor activity of 225-NP in part is mediated by inducing DNA damage in tumor cells. Our study results provide an opportunity for combining 225-NP with other DNA-damaging agents to achieve enhanced antitumor activity.
Materials and methods
Ethics statement
The animal experiments described in this study were carefully reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at MD Anderson Cancer Center, Houston, TX, USA. The study was conducted per institutional IACUC-approved guidelines and ensured proper welfare of the animals. Animals were monitored daily to ensure minimal pain and distress. Health of the animals was monitored by veterinarians, and the cages were kept clean by weekly changing of beds and drinking water. To reduce and minimize pain during treatment, animals were anesthetized using appropriate anesthesia such as isofluorane.
Cell line and cell culture
The human HCC827 lung cancer cell line was cultured in RPMI 1640 medium (Gibco®; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 μg/mL streptomycin, and 100 U/mL penicillin.
Synthesis and conjugation of NPs
Synthesis of iron oxide/gold NPs and the chemistry of conjugating antibodies to the NPs have been previously described.16–18 Briefly, monoclonal anti-EGFR antibody (clone 225, [host mouse; Sigma-Aldrich Co., St Louis, MO, USA]), anti-EGFR antibody (clone 29.1), or anti-rabbit immunoglobulin G (IgG) monoclonal antibody (clone RG-16, Sigma-Aldrich Co.) were used to attach to the gold surface via a linker (SensoPath Technologies, Bozeman, MT, USA) that consisted of a short polyethylene glycol chain terminated at one end by a hydrazide moiety and at the other end by two thiol groups, as previously described.18 The final antibody-conjugated NPs were labeled as 225-NP, 29.1-NP, and IgG-NP and used in the present study. Iron oxide/gold NPs and free unconjugated 225 antibodies were also used in the study and referred to as AuFe and 225-Ab, respectively.
The size and surface charge (zeta potential) of the synthesized NPs were determined as previously described.16 The size of the NPs was 73±35 nm, and they had a surface charge of 29±1 mV, which was in agreement with our previous findings.16
Cell viability assay
HCC827 cells were seeded at 1.0×105 cells/well in six-well plates and incubated overnight at 37°C. The cells were then treated with 3.0×109 particles/mL of AuFe, 29.1-NP, and 225-NP or 0.03 μg/mL of 225-Ab (antibody amount is equivalent to the antibody attached on the surface of 225-NP). At 72 hours after treatment, the cells were harvested by trypsinization and cell viability determined by trypan blue staining as previously described.16,19 The viability of the untreated cells (the control) was considered 100%. The number of viable cells per treatment was determined and expressed as percentage surviving compared with untreated control cells.
Western blotting
Whole cell lysates were collected with RIPA (radioimmunoprecipitation assay) buffer, and protein concentration was calculated by protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA) as previously described.16,19 Total cellular proteins (50 μg) were applied and separated by SDS (sodium dodecyl sulfate) 7.5%–12.5% PAGE (polyacrylamide gel electrophoresis) and transferred electrophoretically to PVDF-Plus membranes (Micron Separations, Westborough, MA, USA). Primary antibodies against PARP (1:1,000 dilution), phospho-Cdc2 (Tyr15; 1:1,000 dilution), phospho-histone H3 (Ser10; D2C8; 1:1,000 dilution; Cell Signaling Technology, Beverly, MA, USA), anti-phospho/total-EGFR antibody (1:500 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA), phospho-histone H2AX (Ser139) (1:2,500 dilution; EMD Millipore, Billerica, MA, USA), BRCA1 (1:5,000 dilution; Novus, Littleton, CO, USA), Chk1 (G-4), and Cdc2 p34 (1:10,000 dilution; Santa Cruz Biotechnology) were purchased and used. LC3B antibody (1:5,000 dilution) was generated as previously described.16,20 β-actin antibody (1:1,000 dilution; Sigma-Aldrich Co.) was used for detecting β-actin expression for ensuring equal protein loading that served as internal loading control. Proteins were detected using appropriate secondary antibodies purchased from commercial vendors and HyGlo ECL reagent (Denville Scientific, Metuchen, NJ, USA) according to the manufacturer’s instructions.
Flow cytometry
Cells that were either not treated (control) or treated with AuFe, 29.1-NP, 225-NP, 225-Ab, or a mixture of AuFe and 225-Ab were collected at 24 and 48 hours after treatment, washed twice with phosphate buffered saline (PBS) (pH 7.2) and fixed with 70% ethanol. The cells were subsequently stained with propidium iodide solution (Hoffman-La Roche Ltd., Basel, Switzerland) and analyzed with a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) as previously described.16,19 The number of cells in the various phases of the cell cycle was determined, and the data were analyzed using the FlowJo software (Tree Star Inc., Ashland, OR, USA).16
Optical imaging
To determine 225-NP uptake by HCC827 cells, we conducted optical imaging studies as previously described.16 Briefly, cells resuspended in phenol-free RPMI medium were seeded in two-well chamber slides and incubated overnight at 37°C, 5% CO2. The following day, the cells were treated with IgG-NP or 225-NP. At 1 hour and 24 hours after treatment, the cells were washed, fixed in 1% paraformaldehyde, and imaged using dark-field reflectance microscopy. All dark-field images were acquired using DM6000 microscope (Leica Microsystems, Wetzlar, Germany) equipped with 20× dark-field objective and Xenon-lamp white-light illumination.
Immunofluorescence staining
HCC827 cells were seeded at 5.0×104 cells/well in Lab-Tek two-well chamber slides (Nalge Nunc International, Rochester, NY, USA) and treated with 3.0×109 particles/mL of AuFe, IgG-NP, and 225-NP or 0.03 μg/mL of 225-Ab. Twenty-four hours later, the cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.5% Triton X for 10 minutes and then incubated with primary antibody against phospho-histone H2AX (Ser139) (1:200 dilution; Cell Signaling Technology) overnight at 4°C. The following day, the cells were washed with PBS buffer (pH 7.2) and stained with Alexa-Fluor 488 goat anti-rabbit IgG (1:5,000 dilution; Thermo Fisher Scientific) for 1 hour and DAPI (4′,6-diamidino-2-phenylindole) for 5 minutes. The slides were cover-slipped and observed with IX81 microscope (Olympus Corporation, Tokyo, Japan). The number of cells positive for histone H2AX foci was determined in a blinded fashion by randomly examining more than 200 cells for each treatment group and subjecting data to statistical analysis. The results thus obtained were represented as the percentage of cells positive for H2AX. The number of H2AX foci per cell per treatment was also determined, and the data were represented in a graphical form.
In vivo subcutaneous tumor model
HCC827 cells (6.0×106 cells/mouse) were injected subcutaneously into the lower right flank of 4–6-week-old female nude mice. When the tumors reached a size of about 5–7 mm in diameter, the mice were divided into four groups (n=8–10/group). Group 1 mice were treated with IgG-NP (8.0×109 particles); group 2 mice were treated with 225-NP (8.0×109 particles); group 3 mice were treated with 225-Ab (0.08 μg; antibody amount is equivalent to the antibody attached on the surface of 225-NP); group 4 mice did not receive any treatment and served as untreated control. Mice were treated twice a week for 3 weeks (six times in total) by intratumoral administration of NPs or free 225-Ab. The perpendicular diameter of each tumor was measured every 3–4 days, and the tumor volume was calculated as previously described21 using the following formula:
tumor volume (mm3) = a × b2 ×0.5
where a is the longest diameter, b is the shortest diameter, and 0.5 is a constant to calculate the volume of an ellipsoid. The data were plotted as average mean tumor volume for each time point for each of the animal groups included in the study.
For determining whether 225-NP inhibited phosphorylated EGFR (pEGFR) and induced apoptosis in vivo during early treatment period, three mice from each group were euthanized on day 10 and the tumors were harvested and snap-frozen and stored at -80°C. The tissues were subsequently used in molecular and immunohistochemistry studies that are described below. All of the animal experiments were conducted under the IACUC-approved guidelines.
Immunohistochemistry
Subcutaneous tumors established in mice as described above for in vivo studies were treated with 225-Ab (n=3), IgG-NP (n=3), or 225-NP (n=3) for three doses (day 0, 4, and 7). Mice were euthanized on day 10, and tumors were harvested for immunohistochemical studies. Tumor tissues were snap-frozen and stored until use. Frozen tumor tissues were sectioned (4–6 μm) and were fixed with 4% paraformaldehyde and permeabilized with protease K solution. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using DeadEnd™ Fluorometric TUNEL System (Promega Corporation, Fitchburg, WI, USA) as per manufacturer recommendations. The stained slides were subsequently observed under IX71 inverted microscope (Olympus). The number of TUNEL-positive cells was counted, and data were represented as the average mean for each treatment group.
Tissue sections were also stained for pEGFR using anti-human pEGFR (Tyr1173) antibody (Cell Signaling Technology). Tissue sections were incubated with pEGFR antibody (1:1,000 dilution) at 4°C overnight. The following day, the tissue sections were washed three times with PBS (pH 7.2) and then incubated with Alexa-Fluor 488 secondary antibody (1:1,000; Thermo Fisher Scientific) at room temperature for 1 hour. Tissue sections were subsequently washed with PBS three times and cover-slipped using aqueous mounting medium. The slides were then observed on IX71 inverted microscope (Olympus), the number of pEGFR-positive cells were counted, and the data were represented as the average mean for each treatment group.
Statistical analysis
All data were expressed as means and 95% confidence intervals. Differences between groups were examined for statistical significance with the Student’s t-test, and P-values <0.05 were considered statistically significant.
Results and discussion
Treatment with 225-NP reduces tumor cell viability through induction of autophagy and apoptosis
Synthesis and physicochemical characterization of the particles were performed as previously described.16 The size of the 225-NPs as determined by dynamic light scattering was 73±35 nm, and they had a surface charge of -29±1 mV as previously described.16
Prior to testing the antitumor activity of 225-NP, we first tested the cellular uptake of the NPs in EGFR-positive HCC827 lung tumor cells and the inhibitory effect of EGFR. Uptake of 225-NP by the tumor cells was detectable as early as 1 hour after NP addition and increased with time, with maximum uptake detected at 24 hours (Figure 1A). Minimal to no NP uptake was observed in cells that were treated with control IgG antibody conjugated-NP at both time points tested. We next determined the 225-NP-mediated inhibitory effects on EGFR expression in HCC827 tumor cells. Reduction in the phosphorylated form of EGFR was observed in 225-NP-treated cells but not in control NP-treated cells (Figure 1B). These results demonstrated 225-NP is specifically taken up by EGFR-positive tumor cells and results in EGFR inhibition, an observation that concurs with our previous report.16
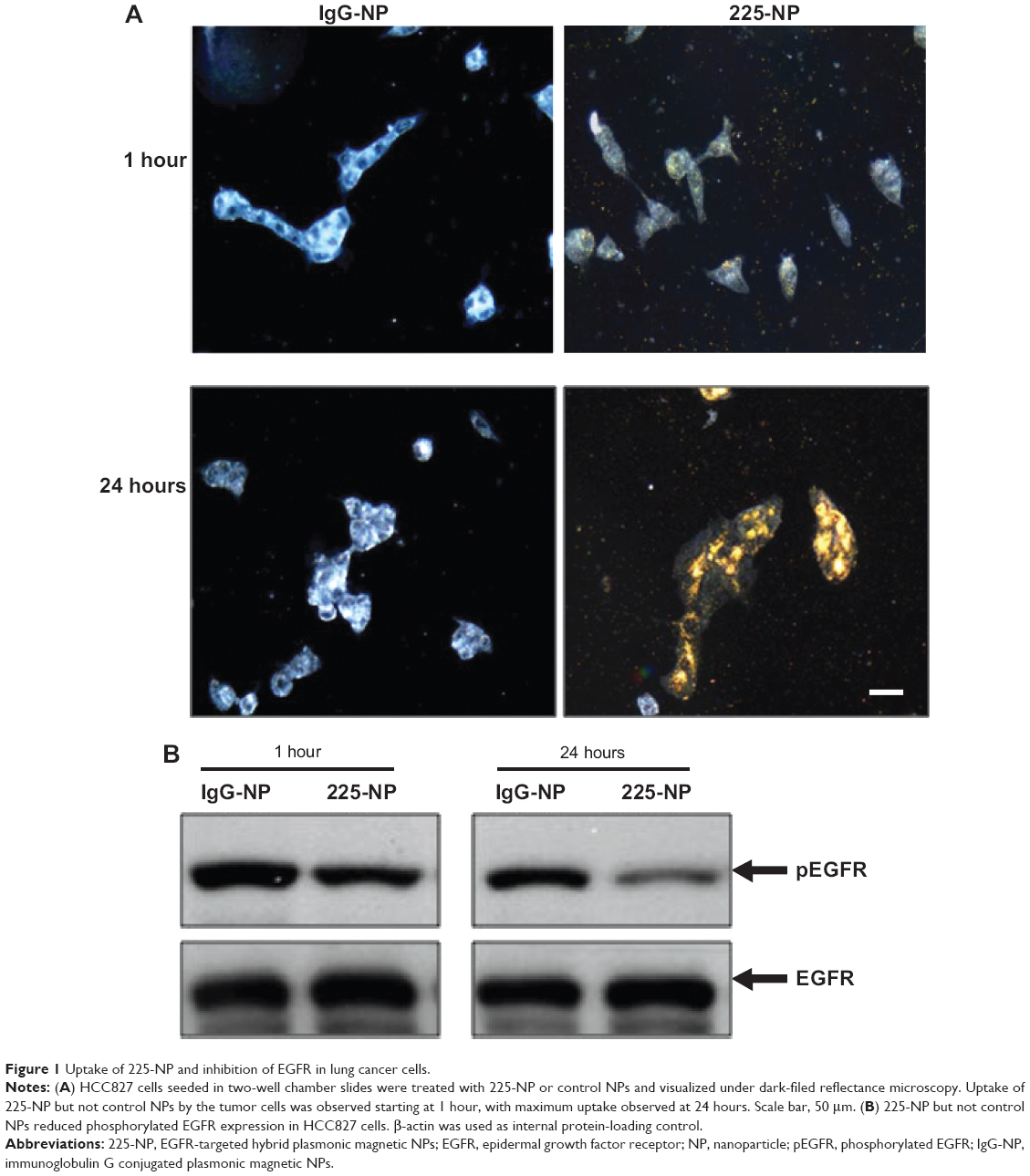 | Figure 1 Uptake of 225-NP and inhibition of EGFR in lung cancer cells. |
To examine the cytotoxic activity of 225-NP on the tumor cells, we performed cell viability assay. Treatment with 225-NP significantly reduced tumor cell viability compared with the cell viability in control groups that were either not treated or treated with control NPs (P<0.05) (Figure 2A). Although both 225 and C29.1 antibodies bind to EGFR, C29.1 antibody unlike 225 binds to a carbohydrate residue on the external portion of EGFR and does not inhibit EGF-mediated EGFR signaling.16,22 Adding a mixture of AuFe and free 225-Ab was much less effective than 225-NP, indicating that the attachment of anti-EGFR antibody on the surface of NPs plays an important role in enhancing the cytotoxic activity of 225-NP (Figure 2A).
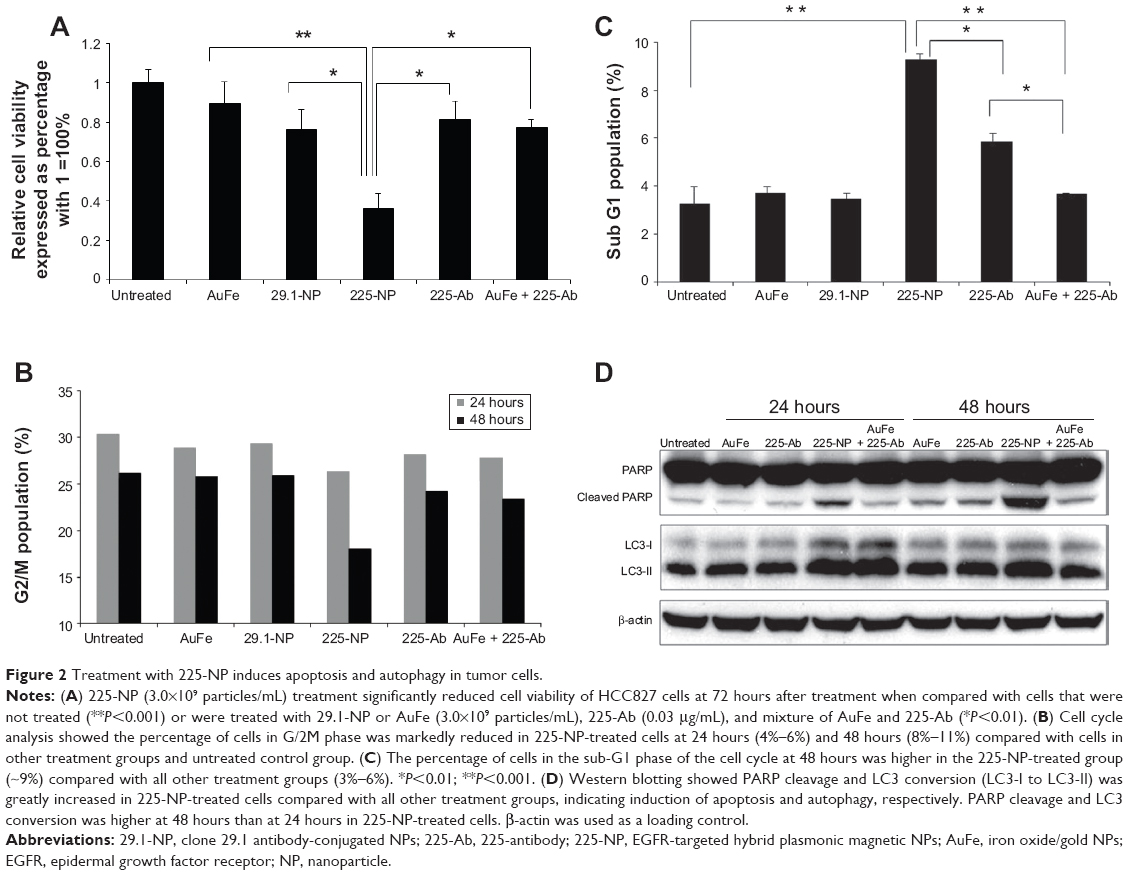 | Figure 2 Treatment with 225-NP induces apoptosis and autophagy in tumor cells. |
Enhanced antitumor activity against human breast cancer cells by combining Her2 antibody to NPs has previously been reported.23 The authors of that study attributed the functional activity to the size of the NPs that correlated with NP uptake by the tumor cells. However, the molecular mechanism of tumor cell killing following uptake of NPs was not demonstrated in that study. Although it is possible NP size could contribute to the antitumor activity, we in the present study did not focus on the size but rather on the mechanism of cell killing.
We next determined the 225-NP treatment effects on cell cycle. Analysis of the G2/M phase by cell cycle analysis showed a decrease in the percentage of cells (~4%–6%) in the G2/M phase at 24 hours after 225-NP treatment compared with the other control treatments (Figure 2B). However, a greater reduction in the percentage of cells (~8%–11%) in the G2/M phase was observed at 48 hours after 225-NP treatment when compared with other treatment groups (Figure 2B). The reduction in G2/M is shown as a range, as the percent difference varied when 225-NP treatment was compared with each individual control. Associated with the decreased G2/M phase was a marked increase in the number of cells in sub-G1 phase in the 225-NP-treated cells (~9%) at 48 hours after treatment compared with the number of cells in the G2/M phase in the control groups (~3%–6%) (Figure 2C). Finally, Western blotting showed increased activation of autophagy and apoptosis as evidenced by very strong LC3 conversion from LC3-I to LC3-II and PARP cleavage, respectively, in 225-NP-treated cells at 24 hours and 48 hours after treatment (Figure 2D). Modest LC3 conversion and PARP cleavage was observed in cells that were treated with free 225-Ab alone at the time points tested (Figure 2D). In AuFe-plus 225-Ab-treated cells, conversion of LC3-I to LC3-II was observed to be equivalent to that observed with 225-NP treatment at 24 hours. However, PARP cleavage was minimal and comparable to untreated control group at 24 hours, suggesting that AuFe plus 225-Ab treatment is able to activate autophagy at 24 hours but unable to activate apoptosis, as evidenced by the reduced PARP cleavage compared with strong PARP cleavage observed with 225-NP treatment. No marked change in LC3 or PARP was observed in other treatment groups when compared with untreated control cells. These results demonstrate 225-NP-mediated cytotoxicity in EGFR-expressing lung cancer cells occurs by a combination of cell-cycle arrest, autophagy, and apoptosis.
An unexpected observation made in our study was that the basal levels of LC3-II in all of the groups was relatively strong over and above which 225-NP treatment showed greater and stronger LC3-II bands. The question that arises is: why are basal LC3-II levels high in the lung cancer cell line used in the present study and what does it mean? The general consensus in the autophagy field is that strong LC3-II band levels represent activation of autophagy while strong LC3-I band levels represent lack of autophagy response. Contrary to the generalization regarding LC3 conversion and autophagy, data exist showing high levels of LC3-II could be detected in untreated control tumor cells. Sato et al24 demonstrated high LC3-II levels in colorectal cancer cell lines and in tumor tissues obtained from patients diagnosed with colorectal cancer and that the high LC3-II levels indicated occurrence of autophagy as a mode for nutrient replenishment and cell survival. Based on that report, we believe that the lung cancer cell line used in the present study likely operates in a fashion similar to that of the colon cancer cell lines, with LC3-II levels being relatively stronger than what is anticipated and supports cell survival. This results in a strong LC3-II level in all of the treatment groups. However, with 225-NP treatment, the autophagy process is greatly enhanced and switched from autophagy to apoptosis, resulting in inhibition of cell growth and induction of apoptotic cell death.
The ability of NPs to induce autophagy and apoptosis has also been demonstrated for other metal and non-metal-based NPs.25–28 However, the majority of these data are from toxicological studies that have focused on investigating NP-mediated toxicity, especially those induced by silica, silver, and zinc oxide-based NPs that are often present in environmental pollutants. The studies were conducted using normal cell lines such as fibroblasts.25–28 Unlike these reports, we have previously demonstrated that 225-NP selectively induces autophagy and apoptosis in lung tumor cell lines but not in normal cell lines.16 Although it is possible, some nonspecific cytotoxicity can occur at very high doses; the doses used in the present study were based on our previous study and were found to selectively kill tumor cells and not normal cells. Thus, we are confident that the observations made in the present study are not related to nonspecific cytotoxicity, as addition of antibody alone or NP alone did not elicit the same degree of cytotoxicity to that observed with 225-NP. Additionally, our study results showed conjugating 225-Ab to the NPs conferred enhanced anticancer activity when compared with activity exhibited by free 225-Ab and NP alone, an observation that concurs with our previous report.16
Although the amount of free 225-Ab added to the cells was equivalent to the amount of 225-Ab attached on the surface of the NPs, the difference in cytotoxic activity observed between 225-NP and 225-Ab suggests that 225-NP in addition to inhibiting the EGFR-tyrosine kinase pathway activates additional cell killing mechanisms, resulting in enhanced cytotoxicity.
Treatment with 225-NP induces DNA damage in tumor cells
To determine the molecular mechanism that contributed to the observed enhanced antitumor activity for 225-NP, we investigated whether 225-NP-mediated tumor cell death involves DNA damage. The rationale to study DNA damage is based on the reports that 225-Ab when combined with radiation therapy results in enhanced DNA damage and radiosensitivity.29,30 Additionally, 225-Ab therapy has been shown to inhibit the movement of the DNA repair enzyme, DNA-dependent protein kinase, from the cytoplasm to the nucleus, thus preventing DNA repair and cell survival.31 Finally, studies are emerging showing NPs can cause DNA damage or DNA damage-like response via oxidative stress to normal cells and thus contribute to toxicity.32–34 On the basis of these reports, we investigated whether 225-NP-mediated antitumor effects involve DNA damage or DNA damage-like response in lung tumor cells by analyzing for phosphorylation of histone H2AX. Histone H2AX becomes phosphorylated at serine 139 in response to DNA double-strand breaks, and phosphorylated H2AX, called γH2AX, is widely used as a DNA damage marker to monitor the effectiveness of cancer therapies.35
Western blotting showed that γH2AX expression level was greatly increased in 225-NP-treated cells compared with the γH2AX expression level in all other treatment groups at both 24 and 48 hours after treatment (Figure 3A). Induction of γH2AX expression, however, was higher at 24 hours than at 48 hours in 225-NP-treated cells. Slight increase in γH2AX expression was observed in cells that were treated with AuFe, 225-Ab alone, and a mixture of NP and 225-Ab when compared with γH2AX expression in untreated cells (Figure 3A). To eliminate the possibility that 225-NP-induced γH2AX expression was simply a phenomenon observed when any antibody is conjugated to the NP, we tested the ability of a control non-therapeutic EGFR antibody (29.1) bound to the NP to induce γH2AX expression. Additionally, we also asked the question of how early after 225-NP treatment γH2AX expression can be observed. Induction of γH2AX expression was observed starting at 24 hours in AuFe-, 29.1-NP-, and 225-NP-treated cells when compared with untreated control cells (Figure S1). However, γH2AX expression was the highest in 225-NP-treated cells compared with 29.1-NP, and AuFe treatment demonstrating γH2AX expression induced by 225-NP was specific and stronger than the expression induced by 29.1-NP. No γH2AX expression was observed in any of the groups at 6 hours after treatment (Figure S1).
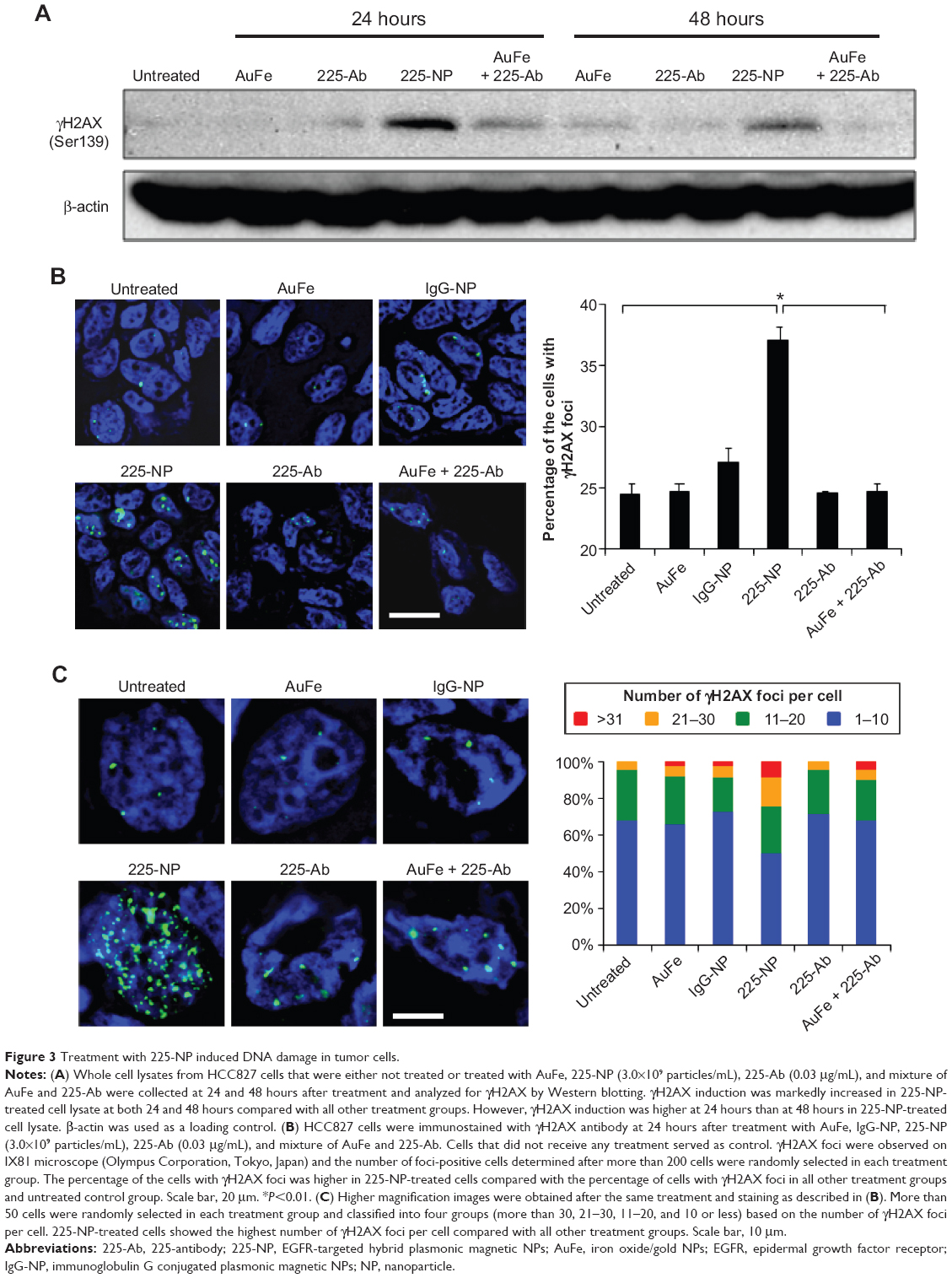 | Figure 3 Treatment with 225-NP induced DNA damage in tumor cells. |
To further determine the location of DNA damage, we performed immunofluorescence staining with γH2AX antibody. Detection of γH2AX staining as foci in the nucleus of a cell is considered an indicator of DNA damage.35,36 Analysis of tumor cells under fluorescence microscopy showed γH2AX foci were in a greater number of cells (~37% of cells) after 225-NP treatment than the number of γH2AX foci observed in cells (~24%–27%) from control treatment groups (Figure 3B). Higher magnification images revealed that 225-NP-treated cells had a larger number of γH2AX foci in the nucleus, and when the number of γH2AX foci per cell was calculated and classified into four groups based on that number, the proportion of the cells with more than 20 foci was higher in 225-NP-treated cells than in the cells from other control groups (Figure 3C). The immunostaining findings with γH2AX showed 225-NP not only induced DNA damage in more cells but induced more DNA damage per cell.
Li et al37 reported gold NPs of 20 nm in size induced oxidative stress that resulted in DNA damage and autophagy in lung fibroblasts. Activation of oxidative stress as measured by reactive oxygen species production has also been reported for other types of NPs.38,39 Whether 225-NP-mediated DNA damage in lung tumor cells was due to oxidative stress was not investigated in the present study. Nevertheless, the ability of 225-NP to induce DNA damage as shown in the present study has potential implication in cancer therapy where 225-NP when combined with other DNA damaging agents such as chemotherapy and radiation therapy is likely to produce enhanced antitumor activity. To our knowledge, this is the first report demonstrating 225-NP in addition to inhibiting EGFR caused DNA damage in lung tumor cells.
Although we have demonstrated 225-NP caused cellular DNA damage, it is of interest to determine the duration of DNA damage following single 225-NP treatment. Knowledge about the DNA repair ability of tumor cells and the time taken to fix the damage will allow an optimal 225-NP-based treatment schedule. Additionally, it will also allow the best possible conditions for combining chemotherapy or radiotherapy for achieving maximal antitumor efficacy. These studies are of interest and are presently being conducted in the laboratory.
The 225-NP abrogates G2/M checkpoint by inhibiting BRCA1-Chk1 pathway
It is widely known that cancer therapeutics such as cisplatin and IR cause DNA damage resulting in the activation of the G2/M checkpoint with the cells arrested in the G2 phase.40,41 This provides sufficient time for the cells to repair the DNA damage and thus prevent the damaged cells from entering mitosis (M phase) with aberrant DNA damage and abnormalities.
DNA damage activates ataxia-teleangiectasia-mutated (ATM) and ATM-Rad3-related signaling and their downstream targets that include breast cancer type 1 susceptibility protein (BRCA1), Chk1, and Chk2 leading to inactivation of Cdc2.42–44 The Cdc2/cyclin B complex plays a pivotal role in regulating G2/M transition. During G2 phase, Cdc2 is inactive when phosphorylated at Tyr15 and Thr14, and dephosphorylation of Cdc2 at Tyr15 and Thr14 by Cdc25c leads the cells to M phase.
Studies have demonstrated NPs, like conventional cancer therapeutics, can induce DNA damage and G2/M cell cycle arrest. Mroz et al45 using a series of NPs that are often found as environmental pollutants showed induction of G2/M arrest and DNA damage in A549 lung cancer cell line when treated with the NPs. Induction of G2/M arrest was accompanied with activation of p53, p53-binding protein (53BP), BRCA1, and γH2AX proteins as determined by increased phosphorylation of these proteins. AshaRani et al46 showed that silver NPs induced DNA damage and produced G2/M cell cycle arrest on IMR-90 (normal human lung fibroblast cells) and U251 (human glioblastoma cells). However, the sensitivity of U251 to silver NPs was higher than the IMR-90 cells, suggesting the tumor cells were more sensitive to the NPs and that using lower doses of silver NPs could be explored for cancer therapy.
On the basis of these reports and our observation of 225-NP treatment reducing the number of cells in the G2/M phase (Figure 2B) and producing DNA damage as indicated by γH2AX foci formation (Figure 3B and C), in the present study, we analyzed for BRCA1 and Chk1 protein expression. As shown in Figure 4, 225-NP markedly reduced both phosphorylated and total BRCA1 and Chk1 protein expression at 24 hours after treatment (Figure 4). Additionally, pCdc2 (Cdk1) and total Cdc2 protein expression, a downstream target of BRCA1–Chk1 pathway was also reduced at 24 hours after 225-NP treatment (Figure 4). Correlating with pCdc2 downregulation was an increase in phosphorylated serine 10 of histone H3, a mitotic marker, at 24 hours after 225-NP treatment. Our findings indicate that 225-NP treatment abrogates the G2/M checkpoint by inhibiting the BRCA1–Chk1 pathway, thus not allowing the cells to efficiently repair the 225-NP-induced DNA damage. As a result, the cells unable to repair the DNA quickly progress to the abnormal mitotic phase, culminating in apoptotic cell death. This possibility is supported by our cell-cycle data showing a reduced number of cells in the G2/M phase along with induction of histone H3 in 225-NP-treated cells, which culminates in an increased number of cells in the sub-G1 and activation of markers related to autophagy and apoptosis. Another line of evidence in support of our findings comes from the study conducted by Yamane et al.47 The authors of the study, using BRCA1-deficient breast cancer cell line (HCC1937), showed partial abrogation of G2/M checkpoint with concomitant failure to inactivate Chk1 via phosphorylation when subjected to IR. In the same study, induction of the mitotic marker, Histone H3 was shown when subjected to IR. This resulted in increased tumor cell apoptosis. However, restoration of wild-type BRCA1 in HCC1937 resulted in inactivation of Chk1 and induction of G2/M cell-cycle arrest when subjected to IR. Thus, BRCA1 plays an important role in cell-cycle and DNA repair, and reduced or loss of BRCA1 expression leads to abrogation of G2/M phase resulting in increased apoptosis.
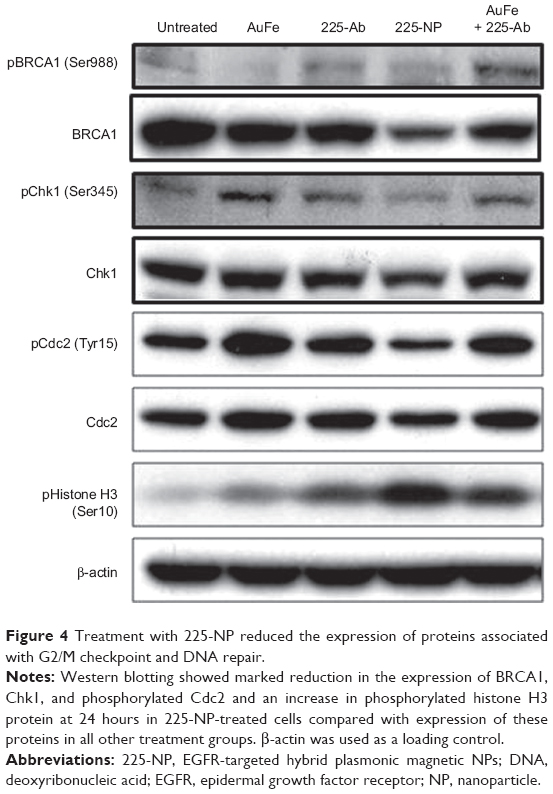 | Figure 4 Treatment with 225-NP reduced the expression of proteins associated with G2/M checkpoint and DNA repair. |
It may be argued that suppression of BRCA1 observed with 225-NP treatment could simply be due to antibody conjugation and is nonspecific. Also, it is important to determine how early after 225-NP treatment BRCA1 gets inhibited. To answer these questions we examined for BRCA1 at 6 and 24 hours post-225-NP treatment and compared with 29.1 antibody-conjugated NP. We observed 225-NP but not 29.1-NP effectively inhibited BRCA1 as early as 6 hours after treatment (Figure S1). Additionally, BRCA1 inhibition appeared to start at 6 hours post-225-NP treatment followed by DNA damage at 24 hours as indicated by induction of γH2AX (Figure S1). These data suggest 225-NP abrogates G2/M early on and is followed by DNA damage resulting in cell death.
Our study results diverge from previous reports in that 225-NP unlike other previously tested NPs did not produce a G2/M arrest but rather reduced the number of cells in G2 phase. Furthermore, we observed reduction in both phosphorylated and total forms of BRCA1, Chk1, and Cdc2 protein expression, which is in sharp contrast to previous reports demonstrated increased expression and activation of these proteins.45,46 Thus, it is evident that the mechanism by which 225-NP produces DNA damage is different from other NPs. One study that concurs with our current findings showed gold NPs induce oxidative stress and DNA damage and reduces BRCA1 mRNA expression.37 However, the authors in that study did not conduct additional molecular studies in vitro or in vivo and did not demonstrate expression of BRCA1 or other cell-cycle proteins being altered at the protein level. Our laboratory is currently investigating the role of oxidative stress and reactive oxygen species in 225-NP-mediated tumor cell killing.
Identification of cell cycle kinases as targets for cancer therapy has resulted in the development and testing of inhibitors targeted to Chk1 and Chk2.44 Combining Chk1 inhibitors with radiation and chemotherapy has demonstrated increased antitumor activity, especially on p53 mutant or deficient cells.48–50 Chk1 and/or Chk2 inhibitors such as UCN-01 and AZD7762 are being tested in Phase I and II clinical trials. Thus, we speculate that 225-NP like Chk1/2 inhibitors will serve as a radiosensitizer and chemosensitizer, producing a greater antitumor effect. However, further investigation is warranted in testing the combinatorial therapy effects, especially in vivo.
Treatment with 225-NP suppresses tumor growth in vivo by inhibiting EGFR phosphorylation and inducing DNA damage
Based on the cytotoxic activity demonstrated by 225-NP toward lung tumor cells in vitro, we investigated the therapeutic efficacy of 225-NP in vivo using a subcutaneous lung tumor xenograft model. Intratumoral administration of 225-NP resulted in a significant inhibition of tumor growth compared with the tumor growth in animals that were untreated or treated with 225-Ab alone or treated with IgG-NP (P<0.05) (Figure 5A). Additionally, tumor growth was observed to be markedly delayed starting from day 14 until day 70 in 225-NP-treated mice when compared with tumor growth in mice from the control groups. In the 225-NP group, three of the mice survived at day 112, while all of the mice in the untreated, 225-Ab-, or IgG-NP-treated control groups were euthanized by day 28 due to tumors reaching a size greater than 1,500 mm3 (Figure 5A).
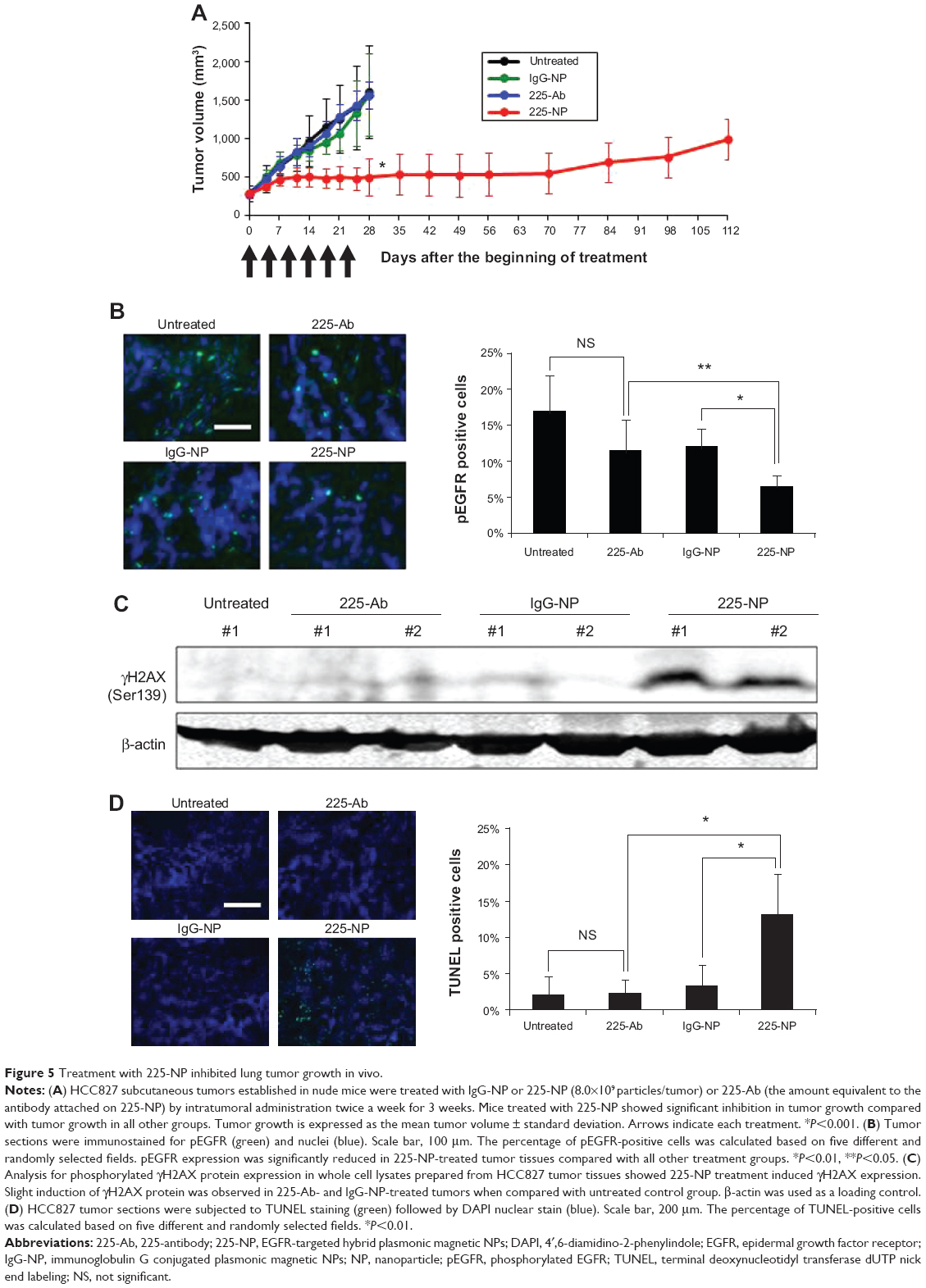 | Figure 5 Treatment with 225-NP inhibited lung tumor growth in vivo. |
We next determined whether 225-NP inhibited EGFR phosphorylation (pEGFR) in vivo akin to the results observed in vitro. Immunohistochemistry studies showed pEGFR expression was significantly inhibited in 225-NP-treated tumor tissues compared with pEGFR expression in 225-Ab- and IgG-NP-treated tumor tissues (P<0.05) (Figure 5B). Although pEGFR expression was reduced in 225-Ab- and IgG-NP-treated tumor tissues compared with pEGFR expression in untreated control tumor tissues, there was no statistical significance (P>0.05). There was no difference in total EGFR expression between tumors treated with 225-NP and the other controls (data not shown).
Since our in vitro studies showed 225-NP treatment induced DNA damage, we determined whether DNA damage occurred in the tumor tissues in vivo. Analysis of total cellular proteins for γH2AX by Western blotting showed strong γH2AX expression in tumors that were harvested from mice treated with 225-NP (Figure 5C). γH2AX expression was detectable in 225-Ab- and IgG-NP-treated tumor tissues, albeit at very low levels when compared with 225-NP-treated tumor tissues. No γH2AX expression was observed in tumor tissues collected from mice that did not receive any treatment.
Finally, TUNEL staining of tumor tissues showed that only 225-NP treatment induced significant tumor cell apoptosis compared with tumor cell apoptosis in all other treatment groups (P<0.05) (Figure 5D). These results demonstrate that 225-NP, in addition to inhibiting pEGFR, induces potent DNA damage, resulting in tumor cell apoptosis and thus producing a strong antitumor activity. Additionally, no observable treatment-related toxicity was observed, indicating 225-NP therapy was safe.
We have, via proof-of-concept studies, demonstrated intratumoral administration of 225-NP is effective in controlling tumor growth. The rationale to first test intratumoral treatment was that it is easy to administer the NPs and monitor and measure tumor growth. However, we are cognizant that for the treatment of metastatic cancer, such as lung cancer, it is important to administer 225-NP intravenously and determine the effectiveness of 225-NP therapy. The systemic treatment approach is more relevant to clinical application and thus warrants testing of intravenous administration of 225-NP in an experimental lung metastasis model. We plan to conduct these studies in the near future. We are also aware that studies testing 225-NP efficacy in additional tumor models, both in vitro and in vivo are warranted. With an increasing number of toxicological study reports demonstrating NPs can cause toxicity to normal tissues, it is also important to determine the biodistribution and toxicity of 225-NP in normal tissues in vivo. In particular, it is important to know whether repeated administration of 225-NPs could lead to accumulation of the particles in the body and unwanted and unexpected chronic toxicity. These studies are important and need to be conducted in the future; the results obtained from these types of studies will provide information that will allow us to make improvements in NP formulation prior to moving forward with clinical testing. An additional and potential challenge in translating 225-NP for clinical testing is in the large-scale manufacturing and quality control, as there is more than one component involved in formulating 225-NP. While it is evident that several questions related to efficacy, toxicity, and clinical translation remain unanswered, these are outside the scope of the present study. Overcoming the hurdles identified above and demonstrating systemic administration of 225-NP is safe and efficacious in controlling primary and metastatic tumors will allow us to advance 225-NP-based therapy for the treatment of patients diagnosed with non-small cell lung cancer.
In summary, we in the present study have established a proof-of-concept and have demonstrated 225-NP has in vivo antitumor efficacy against lung tumors.
Conclusion
In the present study, we have demonstrated 225-NP treatment of EGFR-positive HCC827 lung cancer cells resulted in abrogation of cell cycle arrest in the G2/M phase and induced DNA damage. The consequence of 225-NP treatment is induction of autophagy, apoptosis, and DNA damage that resulted in effective tumor growth inhibition both in vitro and in vivo. Our study has established a proof-of-concept demonstrating a novel mechanism by which 225-NP can produce antitumor activity in lung cancer cells. Although our study results demonstrated 225-NP treatment induced DNA damage in lung tumor cells, testing in additional lung tumor cell lines is warranted. Additionally, the underlying molecular differences in the G2/M checkpoint response to DNA damage caused by 225-NP versus other DNA damaging agents such as IR is yet to be elucidated. Unraveling the mechanism by which 225-NP induces DNA damage will lead to novel innovative combinatorial therapies for lung cancer.
Author contributions
Conducted the experiments, acquired and analyzed data: SK, JT, KS, RR. Synthesized and characterized the NPs: JT and KS. Conceived and designed the experiments: SK, JT, KS, JAR, and RR. Wrote the paper: SK and RR. Reviewed and edited the paper: JT, KS, and JAR. Supervised the project: KS, JAR and RR. All authors critically revised the article and agree to be accountable for all aspects of the work.
Acknowledgments
This study was supported by the National Cancer Institute grants R03EB009182 (RR), R01CA103830 (KS), P50CA070907 (JAR), The Joans Legacy: United against Lung Cancer (RR), and funds from the Jim and Christy Everest Endowed Chair in Translational Cancer Medicine (RR). RR is an Oklahoma TSET Research Scholar and holds the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2000;55:161–171. | ||
Cuenca AG, Jiang H, Hochwald SN, et al. Emerging implications of nanotechnology on cancer diagnostics and therapeutics. Cancer. 2006;107:459–466. | ||
Jain KK. Recent advances in nanooncology. Technol Cancer Res Treat. 2008;7:1–13. | ||
Sokolov K, Follen M, Aaron J, et al. Real-time vital optical imaging of precancer using anti-epidermal growth factor receptor antibodies conjugated to gold nanoparticles. Cancer Res. 2003;63:1999–2004. | ||
Powell AC, Paciotti GF, Libutti SK. Colloidal gold: a novel nanoparticle for targeted cancer therapeutics. Methods Mol Biol. 2010;624:375–384. | ||
Leuschner C, Kumar CS, Hansel W, et al. LHRH-conjugated magnetic iron oxide nanoparticles for detection of breast cancer metastases. Breast Cancer Res Treat. 2006;99:163–176. | ||
Li Z, Kawashita M, Araki N, et al. Preparation of magnetic iron oxide nanoparticles for hyperthermia of cancer in a FeCl2-NaNO3-NaOH aqueous system. J Biomater Appl. 2011;25:643–661. | ||
Veale D, Kerr N, Gibson GJ, Harris AL. Characterization of epidermal growth factor receptor in primary human non-small cell lung cancer. Cancer Res. 1989;49:1313–1317. | ||
Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. | ||
DeGrendele H. Epidermal growth factor receptor inhibitors, gefitinib and erlotinib (Tarceva, OSI-774), in the treatment of bronchioloalveolar carcinoma. Clin Lung Cancer. 2003;5:83–85. | ||
Heymach JV, Nilsson M, Blumenschein G, Papadimitrakopoulou V, Herbst R. Epidermal growth factor receptor inhibitors in development for the treatment of non-small cell lung cancer. Clin Cancer Res. 2006;12:4441s–4445s. | ||
Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. | ||
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and ceuximab plus irinotecan in irinotecan-refractory metatstatic colorectal cancer. N Engl J Med. 2004;351:337–345. | ||
Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. | ||
Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol. 2005;23:2556–2568. | ||
Yokoyama T, Tam J, Kuroda S, et al. EGFR-targeted hybrid plasmonic magnetic nanoparticles synergistically induce autophagy and apoptosis in non-small cell lung cancer cells. PLoS One. 2011;6:e25507. | ||
Lyon JL, Fleming DA, Stone MB, Schiffer P, Williams ME. Synthesis of Fe oxide core/Au shell nanoparticles by iterative hydroxylamine seeding. Nano Letters. 2004;4:719–723. | ||
Kumar S, Aaron J, Sokolov K. Directional conjugation of antibodies to nanoparticles for synthesis of multiplexed optical contrast agents with both delivery and targeting moieties. Nat Protoc. 2008;3:314–320. | ||
Oida Y, Gopalan B, Miyahara R, et al. Inhibition of nuclear factor-kappaB augments antitumor activity of adenovirus-mediated melanoma differentiation-associated gene-7 against lung cancer cells via mitogen-activated protein kinase kinase kinase 1 activation. Mol Cancer Ther. 2007;6:1440–1449. | ||
Aoki H, Kondo Y, Aldape K, et al. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy. 2008;4:467–475. | ||
Ramesh R, Saeki T, Templeton NS, et al. Successful treatment of primary and disseminated human lung cancers by systemic delivery of tumor suppressor genes using an improved liposome vector. Mol Ther. 2001;3:337–350. | ||
Gooi HC, Hounsell EF, Lax I, et al. The carbohydrate specificities of the monoclonal antibodies 29.1, 455 and 3C1B12 to the epidermal growth factor receptor of A431 cells. Biosci Rep. 1985;5:83–94. | ||
Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. | ||
Sato K, Tsuchichara K, Fujii S, et al. Autophagy is activated in colorectral cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–9684. | ||
Li JJ, Hartono D, Ong CN, Bay BH, Yung LY. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31:5996–6003. | ||
Ma X, Wu Y, Jin S, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5:8629–8639. | ||
Zhao Y, Howe JL, Yu Z, et al. Exposure to titanium dioxide nanoparticles induces autophagy in primary human keratinocytes. Small. 2013;9:387–392. | ||
Bexiga MG, Varela JA, Wang F, et al. Cationic nanoparticles induce caspase 3-, 7- and 9-mediated cytotoxicity in a human astrocytoma cell line. Nanotoxicology. 2011;5:557–567. | ||
Wang M, Morsbach F, Sander D, et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011;71:6261–6269. | ||
González JE, Barquinero JF, Lee M, García O, Casaco A. Radiosensitization induced by the anti-epidermal growth factor receptor monoclonal antibodies cetuximab and nimotuzumab in A431 cells. Cancer Biol Ther. 2012;13:71–76. | ||
Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005;76:157–161. | ||
Alarifi S, Ali D, Alkahtani S, et al. Induction of oxidative stress, DNA damage, and apoptosis in a malignant human skin melanoma cell line after exposure to zinc oxide nanoparticles. Int J Nanomedicine. 2013;8:983–993. | ||
Duan J, Yu Y, Li Y, et al. Toxic effect of silica nanoparticles on endothelial cells through DNA damage response via Chk1-dependent G2/M checkpoint. PLoS One. 2013;8:e62087. | ||
Wan R, Mo Y, Feng L, et al. DNA damage caused by metal nanoparticles: involvement of oxidative stress and activation of ATM. Chem Res Toxicol. 2012;25:1402–1411. | ||
Bonner WM, Redon CE, Dickey JS, et al. Gamma H2AX and cancer. Nat Rev Cancer. 2008;8:957–967. | ||
Munshi A, Tanaka T, Hobbs ML, et al. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 2006;5:1967–1974. | ||
Li JJ, Zou L, Hartono D, et al. Gold nanoparticles induce oxidative damage in lung fibroblasts in vitro. Adv Mater. 2008;20:138–142. | ||
Shubayev VI, Pisanic TR 2nd, Jin S. Magnetic nanoparticles for theragnostics. Adv Drug Deliv Rev. 2009;61:467–477. | ||
Singh N, Manshian B, Jenkins GJ, et al. NanoGenotoxicology: the DNA damaging potential of engineered nanomaterials. Biomaterials. 2009;30:3891–3914. | ||
Wang S, Li W, Xue Z, et al. Molecular imaging of p53 signal pathway in lung cancer cell cycle arrest induced by cisplatin. Mol Carcinog. 2013;52:900–907. | ||
Belli M, Sapora O, Tabocchini MA. Molecular targets in cellular response to ionizing radiation and implications in space radiation protection. J Radiat Res. 2002;43 Suppl:S13–S19. | ||
Stark GR, Taylor WR. Control of the G2/M transition. Mol Biotechnol. 2006;32:227–248. | ||
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. | ||
Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. | ||
Mroz RM, Schins RP, Li H, et al. Nanoparticle-driven DNA damage mimics irradiation-related carcinogenesis pathways. Eur Respir J. 2008;31:241–251. | ||
AshaRani PV, Low Kah Mun G, Hande MP, Valiyaveettil S. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano. 2009;3:279–290. | ||
Yamane K, Chen J, Kinsella TJ. Both DNA topoisomerase II-binding protein 1 and BRCA1 regulate the G2-M cell cycle checkpoint. Cancer Res. 2003;63:3049–3053. | ||
Tse AN, Schwartz GK. Potentiation of cytotoxicity of topoisomerase 1 poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–6644. | ||
Koniaras K, Cuddihy AR, Christopoulos H, Hogg A, O’Connell MJ. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene. 2001;20:7453–7463. | ||
Tao Y, Leteur C, Yang C, et al. Radiosensitization by Chir-124, a selective CHK1 inhibitor: effects of p53 and cell cycle checkpoints. Cell Cycle. 2009;8:1196–1205. |
Supplementary material
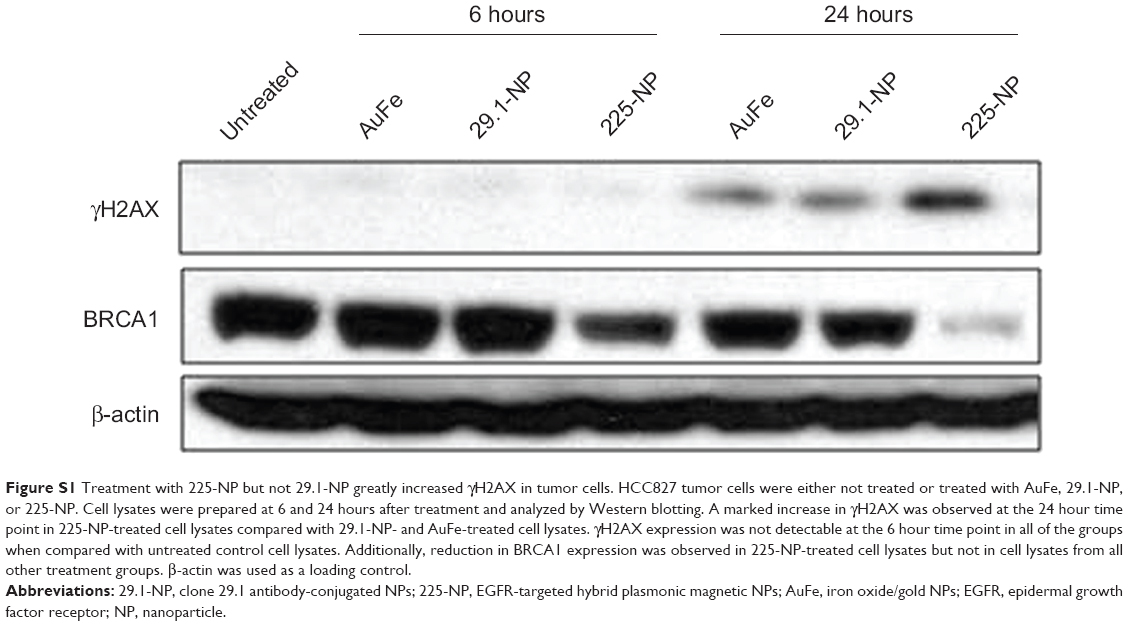 | Figure S1 Treatment with 225-NP but not 29.1-NP greatly increased γH2AX in tumor cells. HCC827 tumor cells were either not treated or treated with AuFe, 29.1-NP, or 225-NP. Cell lysates were prepared at 6 and 24 hours after treatment and analyzed by Western blotting. A marked increase in γH2AX was observed at the 24 hour time point in 225-NP-treated cell lysates compared with 29.1-NP- and AuFe-treated cell lysates. γH2AX expression was not detectable at the 6 hour time point in all of the groups when compared with untreated control cell lysates. Additionally, reduction in BRCA1 expression was observed in 225-NP-treated cell lysates but not in cell lysates from all other treatment groups. β-actin was used as a loading control. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.