")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 8
Dystrophic epidermolysis bullosa: a review
Authors Shinkuma S
Received 14 January 2015
Accepted for publication 31 March 2015
Published 26 May 2015 Volume 2015:8 Pages 275—284
DOI https://doi.org/10.2147/CCID.S54681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Jeffrey Weinberg
Satoru Shinkuma
Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
Abstract: Dystrophic epidermolysis bullosa is a rare inherited blistering disorder caused by mutations in the COL7A1 gene encoding type VII collagen. The deficiency and/or dysfunction of type VII collagen leads to subepidermal blistering immediately below the lamina densa, resulting in mucocutaneous fragility and disease complications such as intractable ulcers, extensive scarring, malnutrition, and malignancy. The disease is usually diagnosed by immunofluorescence mapping and/or transmission electron microscopy and subsequently subclassified into one of 14 subtypes. This review provides practical knowledge on the disease, including new therapeutic strategies.
Keywords: type VII collagen, anchoring fibril, subtypes, revertant mosaicism, treatment, gene therapy
Introduction
Epidermolysis bullosa (EB) is an inherited disorder characterized by mechanical stress-induced blistering of the skin and mucous membranes.1 EB is classified into four major types, namely, EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler syndrome, based on the distinguishing ultrastructural site of skin cleavage.2,3 Tissue separation occurs in the epidermis (EBS), in the lamina lucida (JEB), or in the sublamina densa (DEB). Kindler syndrome, a mixed type, exhibits multiple cleavage planes.
Estimates of the prevalence and incidence of EB have been attempted using different sampling techniques in different regions of the world. There is some variation in the frequency of the DEB among different populations.4–8 According to several registries for EB, the overall prevalence was estimated as 8–10 per million births (for DEB: 2–6 per million births).5,6 The male:female ratio for EB and DEB cases is approximately 1:1. Most DEB patients are symptomatic at birth and until the age of 1 year.
Type VII collagen and the pathogenesis of dystrophic epidermolysis bullosa
DEB is caused by mutations in the COL7A1 gene encoding type VII collagen.9 Type VII collagen is a major component of the anchoring fibril located below the basement membrane in the upper dermis, providing stable dermal–epidermal adhesion.3 Type VII collagen is a homotrimer composed of three proα1 (VII) chains which are encoded by the 32 kb COL7A1 gene on chromosome 3p21.10 The mRNA transcript of approximately 8.9 kb is translated into a procollagen α1 (VII) chain composed of 2,944 amino acids.11 Each procollagen α1 (VII) polypeptide chain contains a central collagenous, triple-helical domain (145 kDa) flanked by both a large amino-terminal noncollagenous 1 (NC-1) domain (145 kDa) and a small carboxyl-terminal NC-2 domain (30 kDa).11,12 The triple-helical domain consists of a repeating Gly-X-Y sequence that is interrupted by 19 noncollagenous regions. In the middle of the triple-helical domain, there is a 39-amino acid noncollagenous region, known as the “hinge” region, which is susceptible to proteolytic digestion with pepsin.13 The amino-terminal NC-1 consists of submodules with homology to known adhesive proteins, including cartilage matrix protein, type III repeats of fibronectin, a von Willebrand factor type A-like motif, and short cysteine- and proline-rich domains.14 The NC-1 domain mediates the attachment of the anchoring fibrils into the principal components of the cutaneous basement membrane, such as type IV collagen and laminin-332.15,16 The carboxyl-terminal NC-2 domain contains conserved cysteines involved in the formation of disulfide bonds which enable linkage between type VII collagen homotrimers.17
Type VII collagen is synthesized both by epidermal keratinocytes and dermal fibroblasts.18 Upon synthesis of a complete procollagen α1 (VII) chain, three polypeptides associate through their carboxyl-terminal ends to form a trimer whose collagenous portion folds into the triple-helical formation.12 The triple-helical molecules are then secreted to the extracellular milieu where two type VII collagen molecules align into a tail-to-tail antiparallel dimer.19 A portion of the NC-2 domain is removed, and the association of the homotrimers is stabilized by intermolecular disulfide bond formation in the overlapping carboxyl-terminal regions. Subsequently, a large number of these antiparallel dimers aggregate laterally to form anchoring fibrils.12
Clinical features of dystrophic epidermolysis bullosa
Clinical subtypes
DEB has been classified according to inheritance pattern, clinical features, and expression level of type VII collagen.1 Four international consensus meetings on EB diagnosis and classification have been held since 1988.1,2,20,21 In the 25 years since the first consensus meeting, several clinical subtypes were identified, and the names of subtypes using eponyms have been eliminated and substituted with descriptive terms. In the most recent proposed classification, there are 14 subtypes of DEB (Table 1).1
 | Table 1 Dystrophic epidermolysis bullosa clinical subtypes |
DEB has two patterns of inheritance: autosomal dominant (DDEB) and autosomal recessive (RDEB). DEB has the three common variants, namely, “DDEB, generalized”, “RDEB, generalized severe”, and “RDEB, generalized intermediate”, as well as other rare variants. “DDEB, pretibial” and “RDEB, pretibial” are rare forms of localized DEB, characterized by recurrent blisters and ulcers and hypertrophic scars occurring mostly in the pretibial area, with variable nail dystrophy.22 “DDEB, pruriginosa” and “RDEB, pruriginosa” are characterized by intense pruritus and nodular prurigo-like legions localized predominantly on the extremities.23 “DDEB, nails only” presents with dystrophic nails, but without skin fragility or trauma-induced blisters.24 “RDEB, inversa” is characterized by generalized blistering from birth and during early infancy.25 Later in infancy or childhood, the predilection sites for blistering change, with the body flexures, axis, and mucous membranes becoming the predominant sites of blistering. “RDEB, centripetalis” is characterized by the slow centripetal progression of symmetrical blister formation and scarring.26 “DDEB, bullous dermolysis of the newborn” and “RDEB, bullous dermolysis of the newborn” present with subepidermal blistering at birth or shortly thereafter, which tends to regress spontaneously in the first few months of life in an autosomal dominant manner and an autosomal recessive manner, respectively.27,28 The clinical features of the three major DEB subtypes are summarized below.
DDEB, generalized
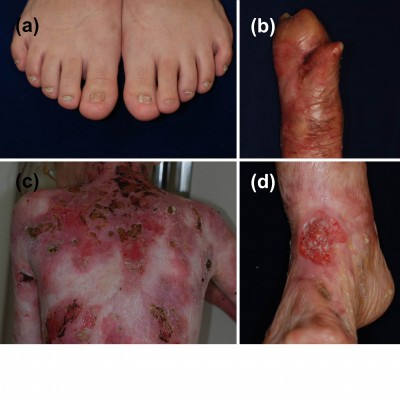
DDEB involves reduced type VII collagen expression and generally has a good prognosis.1,29,30 The blistering is often mild and limited to areas of trauma such as the hands, feet, knees, and elbows, typically resulting in scarring, milia formation, and loss of nails (Figure 1A). Mucosal involvement is rare, and the teeth are normal. The blistering usually starts at birth or soon thereafter, but the disease activity sometimes diminishes with age.
 | Figure 1 Clinical features of dystrophic epidermolysis bullosa. |
RDEB, generalized severe
RDEB, generalized severe is the most severe of all DEB subtypes, because it is caused by absence or marked reduction of type VII collagen expression.1,29,31 The disease, which was previously known as the Hallopeau–Siemens type, presents generalized blisters from birth that result in extensive scarring and pseudosyndactyly and sometime hypo- or hyperpigmentation.29 The hair is typically sparse, with scarring alopecia. Patients develop many problems as a result of the extensive blistering and involvement of the mucous membranes, particularly in the oral cavity, the esophagus, the eyes, and the anal canal.1 Opening of the mouth may become restricted, and the tongue may become less mobile because of scarring. Oral mucosal involvement and esophageal stenosis lead to difficulty in oral feeding.32 Owing to poor nutrition, chronic inflammation, and infection, the patients develop anemia, failure to thrive, delayed puberty, and osteoporosis.33 Eye involvement in DEB patients includes abrasions, scars, and pannus of the cornea; blisters and ectropion of the eyelids; and conjunctival blisters and symblepharon.34 Perianal involvement often leads to painful stools and, as a result, constipation.
RDEB, generalized severe is associated with numerous extracutaneous manifestations including involvement of the gastrointestinal tract, genitourinary tract, kidneys, and heart.35,36 Children with RDEB, severe generalized are at an increased risk of glomerulonephritis, renal amyloidosis, IgA nephropathy, and cardiomyopathy.37,38 Notably, patients have a high likelihood of developing aggressive squamous cell carcinoma (Figure 1D).39 Patients rarely survive beyond age 30, due to severe renal complications or aggressive squamous cell carcinoma.31
RDEB, generalized intermediate
RDEB, generalized intermediate was previously called non-Hallopeau–Siemens type or “RDEB, generalized other”.1,2 The clinical manifestations are similar those of RDEB, generalized severe (Figure 1B and C), but the blistering is less severe, because type VII collagen expression is present, although reduced.29 Patients usually have a better prognosis than that of RDEB, generalized severe. Most patients survive to adulthood, and some are even capable of giving birth. The risk of squamous cell carcinoma is also elevated in this form, and regular follow-up is needed.39
Diagnosis of dystrophic epidermolysis bullosa
After initial diagnosis based on careful examination of the clinical manifestations and inheritance pattern, a skin biopsy from a recently formed blister should be taken to classify the disease by determining the depth of tissue separation.2 Light microscopy can differentiate between intraepidermal and subepidermal split, but it is not very helpful in delineating the specific subtype of EB as both JEB and DEB show subepidermal blistering. Therefore, diagnostic testing and classification in EB begin via immunofluorescence mapping (IFM) and/or transmission electron microscopy (TEM), preferably on fresh blisters.2 Once the level of skin cleavage and the antigen staining profile have been determined, mutational analysis is recommended because this permits the most precise subclassification. Precise diagnosis allows prognostication, genetic counseling, and prenatal diagnosis.9,29
Transmission electron microscopy
TEM and IFM are effective at determining the level of skin cleavage. Currently, IFM is becoming increasingly important in the diagnosis of EB, because TEM requires expensive equipment and significant experience and expertise to process skin biopsy specimens and accurately interpret the resulting micrographs.2 The technique, however, is still useful because TEM allows direct visualization and provides morphologic semiquantitative assessment of structural deficits in the basement membrane zone.3 It also has the advantage of identifying microsplits and subtle changes in the dermal–epidermal junction in DDEB, where IFM results could be normal. Therefore TEM is likely to continue to play an important role in both the clinical and research fields.3
In DEB, subepidermal blistering consistently occurs immediately below the lamina densa (Figure 2). RDEB, generalized severe shows a marked reduction or lack of expression of type VII collagen, which ultrastructurally results in rudimentary or absent anchoring fibrils. In contrast, RDEB, generalized intermediate shows reduced or rudimentary-appearing anchoring fibrils. In DDEB, the anchoring fibrils typically have normal appearance or slightly decreased number.3
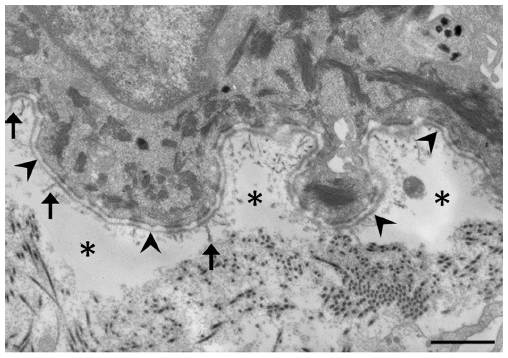 | Figure 2 Electron microscopic image of dystrophic epidermolysis bullosa. |
Immunofluorescence mapping
IFM on a freshly induced blister is now recommended as the primary method for diagnosing EB, because IFM can provide considerable insight into the precise level of tissue cleavage and into the relative expression and distribution of components of the skin basement membrane zone and epidermal antigens.29 As a general rule, the degree of the molecular defect correlates with the clinical severity. In DEB, IFM reveals blister formation below the lamina densa, against which anti-type IV collagen antibodies react.40 Depending on the subtype, the expression level of type VII collagen is varied. In RDEB, generalized severe, type VII collagen is absent or remarkably reduced (Figure 3A). In RDEB, generalized intermediate, type VII collagen usually is reduced to a variable degree (Figure 3B) compared with normal skin (Figure 3C). DEB, bullous dermolysis of the newborn has characteristic IFM findings such as intraepidermal granules of type VII collagen.41,42 Occasionally, IFM fails to provide the exact subtype diagnosis. In the less severe DDEB subtypes, type VII collagen is only slightly reduced and the skin can appear normal by IFM.
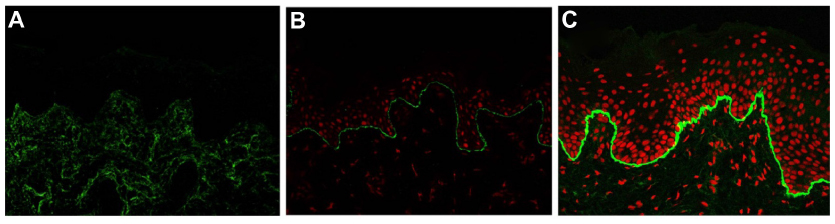 | Figure 3 Immunofluorescence mapping of dystrophic epidermolysis bullosa with antibodies to type VII collagen. |
Mutational analysis
Mutational analysis can provide a definitive diagnosis of EB subtype and mode of inheritance and can determine the precise site and type of molecular mutation in a patient with EB.9,43 Mutation analysis of the COL7A1 gene is performed by using PCR amplification of all 118 exons and exon–intron boundaries and subsequent direct DNA sequencing. Recently, next-generation sequencing has been put to practical use and has allowed a more comprehensive mutation analysis with potential savings of time and money.44
To date, about 400 mutations in COL7A1 have been reported.9,45 In RDEB, compound heterozygosity is common. The different combinations of mutations account for the biologic phenotype, which explains the various clinical severities.29 In most patients with RDEB, generalized severe, premature termination codons (PTC) are present on both alleles of the COL7A1 gene.46 The PTC mutations lead to nonsense-mediated mRNA decay, or residual expression of truncated type VII collagen polypeptides that are degraded within the cells.29 As a result, absent or reduced expression of type VII collagen is observed in the basement membrane in these patients. In RDEB, generalized intermediate, heterogeneous mutations, including missense, in-frame, and splice-site mutations, are present in at least one allele of the COL7A1.29 Frequently, the second mutation causes a PTC. These mutation combinations lead to the synthesis of defective type VII collagen and to structural abnormalities in anchoring fibrils. DDEB usually involves glycine substitutions within the collagenous domain of the collagen α1 (VII) chain in only one allele, although other missense variants, in-frame small insertions or deletions, or splice-site alternations may underlie some cases.30,47 About 1/8 of all type VII collagen trimers (AAA) are normal; 7/8 trimers (AAa, AaA, aAA, Aaa, aAa, aaA, aaa) are disrupted by the abnormal protein. Therefore, the glycine substitutions and the other mutations probably have a dominant negative effect on the formation and assembly of type VII collagen (A: normal type VII collagen, a: mutated type VII collagen).46
Although most COL7A1 mutations have been specific to individual families, some recurrent mutations have been reported in different patients.48 In Italy, the recurrent mutations that were found included insertion/deletion (497insA, 8441-14del21), splice-site (4783-1G>A, 7344G>A), and missense (G1664A) mutations.43 A recurrent frameshift mutation, 2470insG, has been found only in Mexico.49 Recurrent R578X, 7786delG, and R2814X mutations seem to be exclusive to the British population.50,51 The mutations 5818delC, 6573+1G>C, and E2857X have been regarded as recurrent COL7A1 mutations associated with RDEB in Japan.7,48 A high recurrence of 425A>G and G2043R has been found in several different ethnic groups.52
Treatments for patients with dystrophic epidermolysis bullosa
No definitive treatments have been established for DEB, and symptomatic therapies are the mainstay of clinical management.53–55 The prevention of new blisters and ulcers and wound care are the most important aspects of treatment. New blisters should be drained with sterile large-bore needles to prevent extension.55 The blister roof should be left in place, as it acts as a natural wound dressing. Vaseline or other appropriate ointments are first applied to a nonstick dressing, such as Mepilex and Urgotul, and then the lesion is covered with the coated dressing.53,56 The dressing may be fixed with a bandage. Dressings should be changed daily after skin cleansing. Adhesives and compressive dressings must be avoided, because they induce new blisters.
Topical antibiotics and antimicrobial dressings should be used judiciously in DEB patients.56,57 The bacterial colonization of wounds is inevitable, but it often does not interfere with healing.58 In such cases, no treatment is necessary. Due to the increased risk of bacterial resistance, topical antibiotic ointments and antimicrobial dressings should be reserved for those wounds that are colonized with bacteria and fail to heal, referred to as “critical colonization”. Wound infection, which is essentially a clinical diagnosis characterized by increasing size, exudates, odor, pain, surrounding erythema, swelling, and edema, often requires topical or systemic antibiotics. In some cases, topical antibiotics are used on a rotational basis in patients with chronic and/or critically colonized wounds. Silver-impregnated dressings are also effective in reducing the bioburden, but there is a concern about elevated levels of silver in the plasma.59 As noted above, wound management of DEB relies on the recognition of colonization or frank infection.
EB patients have increased caloric and protein needs due to the increased energy expended for wound healing.60 At the same time, involvement of the oropharyngeal, esophageal, and gastrointestinal mucosa in some RDEB patients limits intake, making it difficult to satisfy the high caloric needs. Maximizing nutrition is of vital importance in order to promote growth and development, optimize wound healing, and improve quality of life.
Severe cases with secondary symptoms in different organs require collaboration among pediatricians, dermatologists, internal medicine specialists, dermatologic surgeons, dentists, nutritionists, physical therapists, psychologists, and other specialists.29
Treatment advances
Chronic and recurrent skin ulcers in patients with RDEB cause significant dysfunction, such as contracted scar, and occasionally leads to aggressive squamous cell carcinoma, which often metastasizes and leads to death.39 It is therefore important to achieve early re-epithelialization and prevent recurrent wounds. Various biological dressings, including cultured autologous or allogeneic epidermal and/or dermal grafts, have been used to treat intractable ulcers, and these usually are effective in promoting re-epithelialization.61 The therapeutic effects is temporary, however, and patients experience recurrence at the treated area because the allogeneic grafts provoke immune rejection, and the cultured autografts intrinsically have mutation in COL7A1.62
Allogeneic bone marrow transplantation has been used in the treatment of leukemias, lymphomas, and immune deficiency disorders. It has been shown that bone marrow stem cells have the capacity to migrate to the skin and differentiate into skin cells which produce type VII collagen.63,64 Several groups have conducted bone marrow therapy to treat RDEB-model mice lacking type VII collagen and have confirmed that the treatment can ameliorate the symptoms.63,64 Furthermore, clinical trials of RDEB treatments that use cord blood and bone marrow transplantation have started.65,66 Although these therapies produced phenotypic amelioration, they were also associated with mortality: of the 20 patients with RDEB who received blood and marrow transplantation, five died from disease progression or complications of the transplantation.66
Type VII collagen has been reported to be synthesized and secreted by keratinocytes and fibroblasts. Fibroblasts are much more robust and easier to culture than keratinocytes; therefore, fibroblasts may be better targets for cell-based or ex vivo gene therapy.67 In fact, intradermal injection of normal human fibroblasts or gene-corrected RDEB fibroblasts was found to restore the synthesis and stable deposition of type VII collagen at the dermal–epidermal junction in a murine model.68 A human pilot study of a single intradermal injection of allogeneic fibroblasts was performed in a few individuals with RDEB.69 Although the donor fibroblasts were undetectable at 2 weeks after injection, increased type VII collagen expression was seen at 2 weeks and 3 months in these patients. The investigators suggested that the major effect of allogeneic fibroblast injection is to elevate heparin binding-EGF-like growth factor (HB-EGF) and to increase the recipient’s own COL7A1 mRNA levels that are increased in keratinocytes and fibroblasts.70 Thus, it has been shown that intradermal injections of allogeneic fibroblasts have therapeutic potential in human subjects with RDEB. Intradermal injection of fibroblasts is very painful, and anesthesia may be required. Additional optimization of the frequency of injections and the cell numbers would be necessary for practical application.71
Another attractive approach is recombinant protein therapy. Murine studies have shown that intradermal injections of recombinant human type VII collagen lead to the deposition of type VII collagen at the dermal–epidermal junction.72 Intravenous injection of recombinant type VII collagen into RDEB mice produced systemic deposition of type VII collagen in wounded skin. In addition, not only was topical type VII found to improve the phenotype in an RDEB murine model, but it may also accelerate wound healing in skin.73 About 60–120 mg type VII collagen might be necessary per infusion to translate the results of intravenous injection from a mouse model weighing 35 g injected with 60 μg of type VII collagen recombinant protein to RDEB patients weighing 35–70 kg.74,75 The construction of a large-scale production system and the establishment of product standards would be needed. In addition, pharmacological and toxicology studies using large animals would be mandatory before clinical studies on the safety of the recombinant type VII collagen protein could be conducted.
Because EB is a monogenic disease, corrective gene therapy is an ideal option. Several genetic-based therapeutic approaches have been reported, such as small molecules, ex vivo transfer of the wild-type COL7A1 gene, trans-splicing and homologous directed repair using gene-editing techniques.76–79 Aminoglycosides that can induce PTC readthrough restore functional type VII collagen in keratinocytes obtained from RDEB patients whose disorder is caused by nonsense mutations.76 For ex vivo gene transfer approaches to RDEB, the use of retroviral vectors, lentiviral vectors, and nonviral vectors based on the φ31 bacteriophage integrase system have been developed to transfer full-length COL7A1 complementary DNA (8.9 kb) into RDEB keratinocytes and/or fibroblasts because of the large genome size.80 These approaches are efficient ways to produce functional type VII collagen in cells derived from RDEB patients. In fact, ex vivo COL7A1 gene correction and the grafting of keratinocyte sheets onto RDEB patients is in the early stage of a Phase I trial.71 However, the endogenous correction of mutations in the genome could offer benefits over the use of viral vectors, because oncogenic events might occur due to random insertional mutagenesis of the transgene via viral vectors. In addition, the endogenous correction ensures the physiological transcriptional control of the COL7A1 gene. To overcome this problem, spliceosome-mediated trans-splicing has been developed in which gene repair is done by recombining an endogenous target pre-mRNA and an exogenously delivered RNA molecule called a pre-trans-splicing molecule (PTM). The transferred pre-mRNA, which is shorter than full-length COL7A1 cDNA, is replaced by the wild-type coding sequence from the PTM by trans-splicing into the 3′ or 5′ sequence of the target to generate a new reprogrammed mRNA.77 Recently, it has been reported that engineered site-specific endonucleases in combination with a DNA repair template can replace a mutant allele with a wild-type sequence via homologous recombination repair. Zinc-finger nucleases (ZFNs), transcription activator-like effector proteins (TALENs), or clustered regulatory interspaced shot palindromic repeats (CRISPR)-based RNA guided DNA endonucleases have demonstrated the ability to target specific DNA-binding domains.81 Of note, TALENs have been successfully used to correct COL7A1 gene mutations in primary fibroblasts from patients with RDEB.79
Some RDEB patients acquire healthy skin patches that never blister, where increased type VII collagen expression and restoration of anchoring fibrils are shown.82 In these regions, spontaneous COL7A1 gene correction, or “revertant mosaicism”, is observed on gene analysis, a phenomenon that has been discovered in several inherited skin disorders, including the other type of EB.83 Cell therapy using revertant keratinocytes is ideal because these cells do not need gene correction. Initial attempts, which focused on cultured skin derived from the revertant keratinocytes from a JEB patient, were not successful because the degree of reversion was not maintained in the grafts.84 Punch grafting of revertant skin in a JEB patient with intractable ulcers has led to clinical improvement, and immunofluorescence and DNA sequencing showed maintenance of reversion at the recipient sites.85
Induced pluripotent stem (iPS) cells, which can be generated from patients, acquire the capacity for multipotential differentiation and unlimited self-renewal.86 The iPS cells are able to differentiate not only into keratinocytes and fibroblasts but also into hematopoietic stem cells and mesenchymal stem cells that can home to mucocutaneous blisters and differentiate into keratinocytes and fibroblasts.87–90 RDEB patients usually have mucous lesions that are not readily accessible, and it is difficult to transplant skin grafts and inject cultured cells directly into these areas.91 These stem cells and additional modifications, including local application of recombinant homing signals (such as HMGB1), can complement the use of systemic cell therapy.89 Some groups have already succeeded in generating iPS cells derived from the revertant keratinocytes of EB patients and from fibroblasts corrected with homologous recombination-based gene therapy using TALENs.79,91,92 Therefore, iPS cell therapy using naturally (revertant mosaicism) or artificially (gene editing) gene-treated somatic cells derived from DEB patients is of great promise.
Conclusion
Global patient advocacy groups (DebRA, http://www.debra-international.org/homepage.html) and a clinical network of EB centers and experts (EB-CLINET, http://www.eb-clinet.org/home.html) have been established because the clinical manifestations of EB are so severe. Thus, EB has a high profile not only in the clinical and research fields, but also among the general public. The development of therapies is an urgent issue. In the two decades since the pathogenic mechanism of DEB was clarified, molecular biology and cell biology have dramatically developed, and recent data from animal models, translational research, and initial clinical trials for DEB show great promise. Further research is required in order to realize practical application of effective new therapies.
Acknowledgments
This work was supported by the National Institute of Biomedical Innovation, Japan.
Disclosure
The author reports no conflicts of interest in this work.
References
Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. | |
Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58(6):931–950. | |
Shinkuma S, McMillan JR, Shimizu H. Ultrastructure and molecular pathogenesis of epidermolysis bullosa. Clin Dermatol. 2011;29(4):412–419. | |
McKenna KE, Walsh MY, Bingham EA. Epidermolysis bullosa in Northern Ireland. Br J Dermatol. 1992;127(4):318–321. | |
Kho YC, Rhodes LM, Robertson SJ, et al. Epidemiology of epidermolysis bullosa in the antipodes: the Australasian Epidermolysis Bullosa Registry with a focus on Herlitz junctional epidermolysis bullosa. Arch Dermatol. 2010;146(6):635–640. | |
Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12. | |
Shinkuma S, Natsuga K, Nishie W, Shimizu H. Epidermolysis bullosa in Japan. Dermatol Clin. 2010;28(2):431–432, xvi. | |
Horn HM, Priestley GC, Eady RA, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol. 1997;136(4):560–564. | |
Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17(7):553–568. | |
Ryynanen M, Knowlton RG, Parente MG, Chung LC, Chu ML, Uitto J. Human type VII collagen: genetic linkage of the gene (COL7A1) on chromosome 3 to dominant dystrophic epidermolysis bullosa. Am J Hum Genet. 1991;49(4):797–803. | |
Christiano AM, Greenspan DS, Lee S, Uitto J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J Biol Chem. 1994;269(32):20256–20262. | |
Chung HJ, Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin. 2010; 28(1):93–105. | |
Christiano AM, Hoffman GG, Chung-Honet LC, et al. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics. 1994;21(1):169–179. | |
Christiano AM, Rosenbaum LM, Chung-Honet LC, et al. The large non-collagenous domain (NC-1) of type VII collagen is amino-terminal and chimeric. Homology to cartilage matrix protein, the type III domains of fibronectin and the A domains of von Willebrand factor. Hum Mol Genet. 1992;1(7):475–481. | |
Chen M, Marinkovich MP, Veis A, et al. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J Biol Chem. 1997;272(23):14516–14522. | |
Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC1 domain of collagen VII to laminin 5 and collagen IV. Biochem Biophys Res Commun. 2006;343(3):692–699. | |
Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993;101(3):252–255. | |
Ryynanen J, Sollberg S, Parente MG, Chung LC, Christiano AM, Uitto J. Type VII collagen gene expression by cultured human cells and in fetal skin. Abundant mRNA and protein levels in epidermal keratinocytes. J Clin Invest. 1992;89(1):163–168. | |
Bruckner-Tuderman L, Nilssen O, Zimmermann DR, et al. Immunohistochemical and mutation analyses demonstrate that procollagen VII is processed to collagen VII through removal of the NC-2 domain. J Cell Biol. 1995;131(2):551–559. | |
Fine JD, Bauer EA, Briggaman RA, et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa. A consensus report by the Subcommittee on Diagnosis and Classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol. 1991; 24(1):119–135. | |
Fine JD, Eady RA, Bauer EA, et al. Revised classification system for inherited epidermolysis bullosa: report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42(6):1051–1066. | |
Hamada T, Fukuda S, Ishii N, et al. A Japanese family with dominant pretibial dystrophic epidermolysis bullosa: identification of a new glycine substitution in the triple-helical collagenous domain of type VII collagen. J Dermatol Sci. 2009;54(3):212–214. | |
McGrath JA, Schofield OM, Eady RA. Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features. BrJ Dermatol. 1994;130(5):617–625. | |
Dharma B, Moss C, McGrath JA, Mellerio JE, Ilchyshyn A. Dominant dystrophic epidermolysis bullosa presenting as familial nail dystrophy. Clin Exp Dermatol. 2001;26(1):93–96. | |
van den Akker PC, Mellerio JE, Martinez AE, et al. The inversa type of recessive dystrophic epidermolysis bullosa is caused by specific arginine and glycine substitutions in type VII collagen. J Med Genet. 2011;48(3):160–167. | |
Fine JD, Osment LS, Gay S. Dystrophic epidermolysis bullosa. A new variant characterized by progressive symmetrical centripetal involvement with scarring. Arch Dermatol. 1985;121(8):1014–1017. | |
Hashimoto K, Matsumoto M, Iacobelli D. Transient bullous dermolysis of the newborn. Arch Dermatol. 1985;121(11):1429–1438. | |
Fassihi H, Diba VC, Wessagowit V, et al. Transient bullous dermolysis of the newborn in three generations. Br J Dermatol. 2005;153(5):1058–1063. | |
Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28(1):107–114. | |
Sawamura D, Nizeki H, Miyagawa S, Shinkuma S, Shimizu H. A novel indel COL7A1 mutation 8068del17insGA causes dominant dystrophic epidermolysis bullosa. Br J Dermatol. 2006;154(5):995–997. | |
Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30(1):70–77. | |
Fine JD, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46(2):147–158. | |
Gamelli RL. Nutritional problems of the acute and chronic burn patient. Relevance to epidermolysis bullosa. Arch Dermatol. 1988;124(5):756–759. | |
Figueira EC, Murrell DF, Coroneo MT. Ophthalmic involvement in inherited epidermolysis bullosa. Dermatol Clin. 2010;28(1):143–152. | |
Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61(3):367–384; quiz 385–366. | |
Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61(3):387–402; quiz 403–384. | |
Fine JD, Johnson LB, Weiner M, et al. Inherited epidermolysis bullosa and the risk of death from renal disease: experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis. 2004;44(4):651–660. | |
Fine JD, Hall M, Weiner M, Li KP, Suchindran C. The risk of cardiomyopathy in inherited epidermolysis bullosa. Br J Dermatol. 2008; 159(3):677–682. | |
Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986–2006. J Am Acad Dermatol. 2009;60(2):203–211. | |
Hintner H, Stingl G, Schuler G, et al. Immunofluorescence mapping of antigenic determinants within the dermal-epidermal junction in the mechanobullous diseases. J Invest Dermatol. 1981;76(2):113–118. | |
Fine JD, Johnson LB, Cronce D, et al. Intracytoplasmic retention of type VII collagen and dominant dystrophic epidermolysis bullosa: reversal of defect following cessation of or marked improvement in disease activity. J Invest Dermatol. 1993;101(2):232–236. | |
Radkevich-Brown O, Shwayder T. Bullous dermolysis of the newborn: four new cases and clinical review. Pediatr Dermatol. 2013;30(6):736–740. | |
Gardella R, Castiglia D, Posteraro P, et al. Genotype-phenotype correlation in Italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2002;119(6):1456–1462. | |
Takeichi T, Nanda A, Liu L, et al. Impact of next generation sequencing on diagnostics in a genetic skin disease clinic. Exp Dermatol. 2013;22(12):825–831. | |
van den Akker PC, Jonkman MF, Rengaw T, et al. The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat. 2011;32(10):1100–1107. | |
Sawamura D, Nakano H, Matsuzaki Y. Overview of epidermolysis bullosa. J Dermatol. 2010;37(3):214–219. | |
Toyonaga E, Nishie W, Komine M, et al. Skipped exon in COL7A1 determines the clinical phenotypes of dystrophic epidermolysis bullosa. Br J Dermatol. 2015;172(4)1141–1144. | |
Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res. 2004;295(10):442–447. | |
Mellerio JE, Salas-Alanis JC, Amaya-Guerra M, et al. A recurrent frameshift mutation in exon 19 of the type VII collagen gene (COL7A1) in Mexican patients with recessive dystrophic epidermolysis bullosa. Exp Dermatol. 1999;8(1):22–29. | |
Mellerio JE, Dunnill MG, Allison W, et al. Recurrent mutations in the type VII collagen gene (COL7A1) in patients with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 1997;109(2):246–249. | |
Mohammedi R, Mellerio JE, Ashton GH, Eady RA, McGrath JA. A recurrent COL7A1 mutation, R2814X, in British patients with recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol. 1999;24(1):37–39. | |
Mellerio JE, Salas-Alanis JC, Talamantes ML, et al. A recurrent glycine substitution mutation, G2043R, in the type VII collagen gene (COL7A1) in dominant dystrophic epidermolysis bullosa. Br J Dermatol. 1998;139(4):730–737. | |
Denyer JE. Wound management for children with epidermolysis bullosa. Dermatol Clin. 2010;28(2):257–264, viii–ix. | |
Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34(1):20–25. | |
Pope E, Lara-Corrales I, Mellerio J, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67(5):904–917. | |
Gonzalez ME. Evaluation and treatment of the newborn with epidermolysis bullosa. Semin Perinatol. 2013;37(1):32–39. | |
Mellerio JE. Infection and colonization in epidermolysis bullosa. Dermatol Clin. 2010;28(2):267–269, ix. | |
van der Kooi-Pol MM, Duipmans JC, Jonkman MF, van Dijl JM. Host-pathogen interactions in epidermolysis bullosa patients colonized with Staphylococcus aureus. Int J Med Microbiol. 2014;304(2):195–203. | |
Flohr C, Heague J, Leach I, English J. Topical silver sulfadiazine-induced systemic argyria in a patient with severe generalized dystrophic epidermolysis bullosa. Br J Dermatol. 2008;159(3):740–741. | |
Haynes L. Nutrition for children with epidermolysis bullosa. Dermatol Clin. 2010;28(2):289–301, x. | |
Shinkuma S, Sawamura D, Fujita Y, et al. Long-term follow-up of cultured epidermal autograft in a patient with recessive dystrophic epidermolysis bullosa. Acta Derm Venereol. 2014;94(1):98–99. | |
Wollina U, Konrad H, Fischer T. Recessive epidermolysis bullosa dystrophicans (Hallopeau-Siemens) – improvement of wound healing by autologous epidermal grafts on an esterified hyaluronic acid membrane. J Dermatol. 2001;28(4):217–220. | |
Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173(3):803–814. | |
Tolar J, Ishida-Yamamoto A, Riddle M, et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113(5):1167–1174. | |
Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363(7):629–639. | |
Tolar J, Wagner JE. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet. 2013;382(9899):1214–1223. | |
Goto M, Sawamura D, Ito K, et al. Fibroblasts show more potential as target cells than keratinocytes in COL7A1 gene therapy of dystrophic epidermolysis bullosa. J Invest Dermatol. 2006;126(4):766–772. | |
Woodley DT, Remington J, Huang Y, et al. Intravenously injected human fibroblasts home to skin wounds, deliver type VII collagen, and promote wound healing. Mol Ther. 2007;15(3):628–635. | |
Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008; 128(9):2179–2189. | |
Nagy N, Almaani N, Tanaka A, et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis Bullosa. J Invest Dermatol. 2011;131(8):1771–1774. | |
Hsu CK, Wang SP, Lee JY, McGrath JA. Treatment of hereditary epidermolysis bullosa: updates and future prospects. Am J Clin Dermatol. 2014;15(1):1–6. | |
Woodley DT, Keene DR, Atha T, et al. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nat Med. 2004;10(7):693–695. | |
Wang X, Ghasri P, Amir M, et al. Topical application of recombinant type VII collagen incorporates into the dermal-epidermal junction and promotes wound closure. Mol Ther. 2013;21(7):1335–1344. | |
Woodley DT, Wang X, Amir M, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133(7):1910–1913. | |
Hovnanian A. Systemic protein therapy for recessive dystrophic epidermolysis bullosa: how far are we from clinical translation? J Invest Dermatol. 2013;133(7):1719–1721. | |
Cogan J, Weinstein J, Wang X, et al. Aminoglycosides restore full-length type VII collagen by overcoming premature termination codons: therapeutic implications for dystrophic epidermolysis bullosa. Mol Ther. 2014;22(10):1741–1752. | |
Murauer EM, Gache Y, Gratz IK, et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131(1):74–83. | |
Chen M, Kasahara N, Keene DR, et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat Genet. 2002;32(4):670–675. | |
Osborn MJ, Starker CG, McElroy AN, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21(6):1151–1159. | |
Vanden Oever MJ, Tolar J. Advances in understanding and treating dystrophic epidermolysis bullosa. F1000Prime Rep. 2014;6:35. | |
Li HL, Nakano T, Hotta A. Genetic correction using engineered nucleases for gene therapy applications. Dev Growth Differ. 2014;56(1):63–77. | |
Almaani N, Nagy N, Liu L, et al. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130(7):1937–1940. | |
Jonkman MF, Scheffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997; 88(4):543–551. | |
Gostynski A, Deviaene FC, Pasmooij AM, Pas HH, Jonkman MF. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br J Dermatol. 2009;161(2):444–447. | |
Gostynski A, Pasmooij AM, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70(1):98–101. | |
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. | |
Frobel J, Hemeda H, Lenz M, et al. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014;3(3):414–422. | |
Itoh M, Kiuru M, Cairo MS, Christiano AM. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2011;108(21):8797–8802. | |
Tamai K, Yamazaki T, Chino T, et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci U S A. 2011;108(16):6609–6614. | |
Itoh M, Umegaki-Arao N, Guo Z, Liu L, Higgins CA, Christiano AM. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PloS One. 2013;8(10):e77673. | |
Tolar J, McGrath JA, Xia L, et al. Patient-specific naturally gene-reverted induced pluripotent stem cells in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2014;134(5):1246–1254. | |
Umegaki-Arao N, Pasmooij AM, Itoh M, et al. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci Transl Med. 2014;6(264):264ra164. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.