")
Back to Journals » The Application of Clinical Genetics » Volume 7
Disease-modifying genes and monogenic disorders: experience in cystic fibrosis
Authors Gallati S
Received 7 March 2014
Accepted for publication 10 May 2014
Published 10 July 2014 Volume 2014:7 Pages 133—146
DOI https://doi.org/10.2147/TACG.S18675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sabina Gallati
Division of Human Genetics, Department of Pediatrics, and Department of Clinical Research, Inselspital, University of Berne, Berne, Switzerland
Abstract: The mechanisms responsible for the determination of phenotypes are still not well understood; however, it has become apparent that modifier genes must play a considerable role in the phenotypic heterogeneity of Mendelian disorders. Significant advances in genetic technologies and molecular medicine allow huge amounts of information to be generated from individual samples within a reasonable time frame. This review focuses on the role of modifier genes using the example of cystic fibrosis, the most common lethal autosomal recessive disorder in the white population, and discusses the advantages and limitations of candidate gene approaches versus genome-wide association studies. Moreover, the implications of modifier gene research for other monogenic disorders, as well as its significance for diagnostic, prognostic, and therapeutic approaches are summarized. Increasing insight into modifying mechanisms opens up new perspectives, dispelling the idea of genetic disorders being caused by one single gene.
Keywords: modifier genes, cystic fibrosis, genome wide association studies, candidate gene approach, genotype, phenotype
Introduction
The mechanisms responsible for the determination and fine-tuning of phenotypes are still not well understood, albeit it is widely accepted that the specific disease genotype, epigenetic factors, and environmental influences are significantly involved. However, it has become increasingly apparent that modifier genes must play a considerable role in the phenotypic heterogeneity of Mendelian disorders, reflected for instance by intrafamilial variations, incomplete penetrance, and varying severity of the clinical course. New genetic technologies and advances in molecular medicine allow huge amounts of information to be generated from individual samples in the form of genomic, transcriptomic, and proteomic data. However, analysis and interpretation of this flood of findings, most notably putting them into the right biological context, represents one of the main challenges for research and medicine today. The purpose of this review is to provide insight into the role of modifier genes as an important source of phenotypic variability in so-called monogenic disorders, using the example of cystic fibrosis (CF). Further, it discusses the benefits and limitations of the actual knowledge as well as future technical and strategic approaches. However, since a comprehensive discussion of all modifier gene studies is beyond the scope of this review, the main focus will rather be on recent progress made in the field.
Modifier genes
Determination of the genotype of a single locus is usually not sufficient to predict a clinical phenotype in human disorders considered to be inherited in simple Mendelian patterns, assuming no clear distinction between monogenic and complex traits or involvement of other molecular mechanisms. Thus, the traditional view of genetic disorders being either monogenic or multifactorial is no longer supported and requires a change in focus towards factors that modify gene expression and disease course (Figure 1). Functioning in a common cellular environment, genes are expected to interact with and influence each other. The interaction between genes at different loci in a such way that they combine to change the disease phenotype is called epistasis,1 and the genes involved are genetic modifiers. Nowadays, it is well recognized that epistasis is a ubiquitous component of human disease and that complex interactions play a more significant role than the independent main effects of any single susceptibility gene.2 Modifier genes can influence the phenotypic outcome of a given genotype via the same, a related, or a parallel biological pathway as the disease gene.3,4 Moreover, for any given disorder, multiple modifier genes may contribute to the phenotype in a cumulative way and have the potential to alter penetrance, expressivity, pleiotropy, and severity, including age of disease onset and progression rate. Modifier genes can enhance the primary defect (synergistic epistasis), leading to a more severe phenotype, or reduce it (antagonistic epistasis) and thus have a protective effect. However, the search for modifier genes is very challenging and disease associations are difficult to establish, because the contribution of a single modifier to the phenotype may be small and can be masked by other influencing factors, such as environment and ethnicity, as well as individual genetic background. Thus, no single genetic approach meets all conditions required for comprehensive and successful identification of modifier genes in monogenic diseases, and the combination of classical approaches with genome-wide screenings may offer the best strategy.
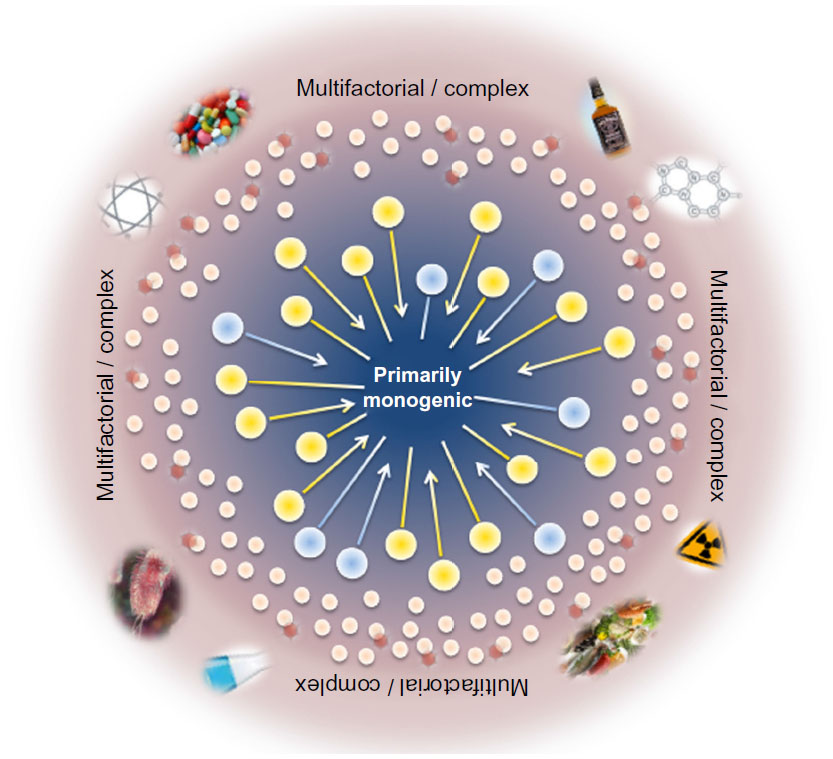 | Figure 1 Scheme of the relationship between genotype and phenotype, and the influence of additional factors such as modifier genes, entire genetic background, epigenetic, and environmental effects on cystic fibrosis. |
Identification of modifier genes
Different mechanisms might explain the variability of monogenic disease expression such as genetic heterogeneity, additional variant(s) in the disease gene in cis or trans position to the primary mutation(s), modifier genes, epigenetic changes (DNA methylation, histone modification), and environmental conditioning. Searching for modifier genes requires a different approach than looking for genes primarily involved in development of a disease. The first and most important step is to define the clinical phenotype of a disease that represents the variability between affected individuals and is expected to be modified by factors other than the primary defect. The second challenge is then to choose the corresponding patient cohort. Strategies to identify modifier genes depend on the data available, and include linkage analyses as well as association studies.
Approaches are either hypothesis-driven, focusing on specific candidate genes and candidate pathways, or represent a blind search systematically scanning the whole genome. The use of single nucleotide polymorphism (SNP) arrays including more than one million markers randomly chosen to cover the genome recently allowed genome-wide association (GWA) studies to be performed in complex and monogenic diseases; however, to obtain the statistical evidence needed to prove an association between a sequence variant and a disease phenotype, very large samples of patients and control individuals are required, a condition that is very hard to fulfill.5 Therefore, for many studies, choosing the candidate gene approach will be more feasible and effective. Candidate genes may be part of the interactome of the primary disease gene or, alternatively, belong to another pathway that is indirectly involved in the disease course (eg, the inflammatory pathway in CF). Thus, resequencing of the selected genes in a carefully characterized patient cohort with well defined phenotypes remains a promising strategy. In summary, the challenge for studying monogenic disorders is to assess the relative contribution of modifier genes to the disease phenotype and to reliably identify these genes, bearing in mind that multiple modifier genes may create a cumulative effect on the phenotypic expression and that the combination of these genes may vary between individuals.
As one of the most common Mendelian disorders and presenting with a broad clinical heterogeneity, CF is one of the best examples to demonstrate strategic approaches for the identification of modifier genes and to explain their involvement in phenotypic expression.
Cystic fibrosis
CF is the most common lethal autosomal recessive disorder in the white population, with an incidence of one in around 2,500 and a carrier frequency of one in 25. Thus, in every 625th couple, both partners are carriers, facing a one in four risk of having a child with CF. The main clinical symptoms of CF are elevated sweat chloride concentrations, exocrine pancreatic insufficiency (in 85%–90% of patients), male infertility, and progressive obstructive lung disease.6,7 Nowadays, lung disease is the major cause of morbidity and mortality in CF, arising from chronic bacterial infections with a persistent neutrophilic inflammatory response and leading to airway damage, bronchiectasis, emphysema, and finally to respiratory failure.8 Additional complications of CF include meconium ileus in the neonatal period, affecting about 15% of newborns with CF,9 severe hepatic cirrhosis and portal hypertension in 3%–5% of patients, and CF-related diabetes, that develops in only 2% of children but in 19% of adolescents and over 40% of adults with CF.10 Pancreatitis is also a well known albeit less common manifestation, that occurs in 15%–20% of pancreatic-sufficient CF patients.11 Moreover, nearly all patients present with sinus disease (chronic sinusitis and/or nasal polyposis), but not all are symptomatic.12 During recent decades, the life expectancy for patients with CF has improved significantly, from less than one year to more than 35 years, due to specific treatments, including pancreatic enzyme and vitamin supplementation, antibiotics, and inhaled bronchodilators and steroids, as well as autogenic drainage and aerobic exercise, indicating a considerable effect of nongenetic environmental factors on disease variability.
In addition to the patients with the classical life-shortening form of CF, there are individuals with cystic fibrosis transmembrane conductance regulator (CFTR)-related metabolic syndrome or with monosymptomatic disorders, such as congenital bilateral absence of the vas deferens, acute recurrent or chronic pancreatitis, and disseminated bronchiectasis. This clinical entity of disorders that do not meet the diagnostic criteria of CF but are associated with CFTR dysfunction is classified as CFTR-related disorders.13
CFTR gene and protein
The gene causing CF, identified in 1989,14,15 is located on chromosome 7q31.2, spanning a transcription unit of ~216.7 kb and containing 27 exons.16 It is transcribed into 6.1 kb messenger ribonucleic acids (mRNAs) encoding the transmembrane CFTR protein of 1,480 amino acids. The main function of CFTR is that of a cyclic AMP-regulated chloride channel located at the apical membrane of polarized epithelial cells that is expressed in several functionally diverse tissues, including the lungs, sweat ducts, pancreas, gastrointestinal tract, and vas deferens.17,18 Moreover, CFTR directly mediates secretion of bicarbonate across the apical membrane linking ion transport and luminal pH, with mucin secretion, plugging, and retention, which are the hallmarks of CF pathology.19–21 CFTR-mediated chloride secretion across epithelial cells is controlled by both modulating channel activity and regulating the total number of CFTR channels in the membrane.22 Alterations in the synthesis or sequence of the CFTR protein may affect the number of channels in the plasma membrane, channel activity, and/or intracellular trafficking of CFTR.
Genotypic and phenotypic heterogeneity
Nearly 2,000 different sequence variants have been reported so far to The Cystic Fibrosis Genetic Analysis Consortium (CFTR1 database, http://www.genet.sickkids.on.ca/cftr/app). The most common defect causing CF is the F508del mutation (c.1521_1523delCTT; p.Phe508del according to the current standard nomenclature), a 3 bp deletion in exon 10 causing loss of the amino acid phenylalanine at position 508 of the protein.23 Worldwide, this allele accounts for ~60% of all CF chromosomes (range 27%–87.5%), with considerably variable frequencies depending on populations and geographic locations.24 There are another 23 relatively common mutations (frequency >0.5%) worldwide and a few mutations with an unusually high frequency in specific populations, indicating a founder effect and genetic drift. The remaining mutations represent rare or even individual alleles that are distributed throughout the entire gene. Mutations (missense, nonsense, frameshift, splice, small and large in-frame deletions and insertions) contribute to the phenotype by their nature and position in the gene and can be classified by their molecular effects on CFTR.25,26 Class 1 mutations are defined as loss of function, and thus are expected to result in no functional CFTR protein and to be associated with a severe phenotype. Class 2 mutations produce misprocessed/misfolded proteins (eg, F508del), class 3 mutations affect activation or gating of the CFTR channel (eg, G551D), class 4 mutations reduce ion conductance through the channel pore (eg, R117H, R334W, R347P), and class 5 mutations reduce the amount of functional CFTR either by partially aberrant splicing (eg, 3849 +10kbC>T, 5T) or by inefficient trafficking (eg, A455E). Class 6 includes nonsense and frameshift mutations (eg, Q1412X, 4326delTC, 4279insA) causing a 70–100 bp truncation of the C-terminus of the CFTR that leads to a marked instability of an otherwise fully processed and functional protein and as a consequence to a severe CF presentation.27 Mutations in any of the classes 2–5 display a broad range of phenotypic effects, making prediction of the clinical course of a patient impossible simply based on mutation classification. Moreover, the potential of a mutation to contribute to the phenotype depends not only on its nature, localization in the gene, and molecular mechanism, but also on its interaction with the second mutated CFTR allele and on disease modifiers. Although CF is considered a monogenic disorder, studies of clinical phenotype in correlation with genotype have revealed a very complex relationship. Some phenotypic features are closely determined by the genotype in an essentially monogenic fashion, whereas others are strongly influenced by both modifying genetic factors and the environment. There is a close relationship between the CFTR genotype and the pancreatic phenotype, revealing “severe” mutations (eg, F508del, all class 1 mutations) to be associated with pancreatic insufficiency and “mild” mutations, such as a series of missense and alternative splice mutations, to be associated with pancreatic sufficiency.28,29 Interestingly, pancreatic-sufficient patients carrying two mild CFTR mutations are at significantly higher risk of developing pancreatitis than patients with moderate or moderate/severe genotypes correlating with the residual CFTR function and the pancreatic acinar reserve.11 On the other hand, because of its complexity and patient exposure to a multitude of endogenous and exogenous factors, the pulmonary outcome is clinically the most variable as well as the most unpredictable component of the CF phenotype. Studies focusing on pulmonary status as a function of the F508del allele have reported a wide range of effects, from a detectable impact of CFTR genotype to no or statistically insignificant influences.30,31 Other studies using more refined assessment of CFTR mutations have shown statistically significant correlations between CFTR genotypes and pulmonary status,32 whereas still others have failed to detect a significant association.33 From my own studies, we can conclude that there is a significant association between specific genotypes and severity of pulmonary disease, and that F508del homozygous patients show the most considerable variation in severity of pulmonary disease.34–36 The impact of CFTR genotypes has also been investigated in other organs, such as the sweat glands and the reproductive tract in men.37,38 One major conclusion resulting from these studies was that expression of CFTR in different organ systems of the same individual is highly variable, and that therefore each organ may be variably affected, making diagnostic and therapeutic approaches much more complicated.
Despite all efforts, the clinical and functional relevance of most CFTR mutations has not been described so far. However, to address this gap, a genotype-phenotype database (http://www.cftr2.org) has been established including nearly 39,700 CF patients, that comprises around 190 CFTR mutations and gives evidence whether a mutation can be classified as causing CF disease, not causing CF, or if varying clinical consequences can be expected. This is an ongoing project and an important addition to the traditional CFTR1 database.
Taken together, all these findings point to the understanding that a CFTR genotype constitutes only the source or potential for CF disease, which will to varying degrees be expressed and translated into CF pathophysiology. Thus, classic genotype–phenotype studies are important but not sufficient and have to be complemented by the search for environmental and non-CFTR genetic effects on clinical phenotype in order to enhance the knowledge of disease mechanisms and to develop new diagnostic and therapeutic approaches.
Role of genetic modifiers in CF
Previous findings indicating a strong influence of genetic modulators on CF phenotype39–41 and identification of macromolecular complexes regulating the intracellular trafficking and activity of CFTR42,43 emphasize the need to focus on the detection of the genes and biological mechanisms involved and provide the well founded basis for extended GWA and candidate gene studies. Two different approaches have been used to identify modifiers in CF, ie, case-control association studies on the one hand and family-based linkage studies on the other. As both methods have their advantages and disadvantages, eg, higher robustness regarding detection of common genetic variants (case-control association studies) or better differentiation between environmental versus genetic effects (family-based studies), complementary studies employing both methodologies are highly recommended.44 Moreover, any association between a chromosomal region, a specific gene locus, or a single gene variant should, if ever possible, be replicated in one or more separate patient and control cohorts,10 although differences in ethnic background, treatment procedures, environmental factors, and socioeconomic influences may weaken or even hide any significant findings.
Genome-wide approaches
CF lung disease
The North American Cystic Fibrosis Gene Modifier Consortium performed a combined GWA and linkage study in a total of 3,467 CF patients and identified significant and replicated association with loci within the chromosomal regions 11p13 and 20q13.2 modifying lung disease severity and harboring genes of biological relevance for CF.45 The strongest signal was located at 11p13, in the intergenic region 3′ to the genes APIP (APAF1-interacting protein) and EHF (ETS homologous factor) with regulatory features and correlation of expression of nearby genes such as EHF to ELF5, EHF to APIP and APIP to PDHX. The EHF gene encodes a protein that belongs to an ETS transcription factor subfamily characterized by epithelial-specific expression and acts as an important regulator of epithelial differentiation under stress conditions and inflammation. Very recently, EHF has been confirmed as a modifier of the CF phenotype in the patient cohort of the European CF Twin and Sibling Study by altering capabilities of the epithelial cells to correctly process folding and trafficking of mutant CFTR.46 The second gene within the 11p13 region, APIP, inhibits cytochrome c-dependent and APFA1-mediated cell death and prevents ischemic/hypoxic injury. It is suggested that increased expression of APIP resulting in exaggerated inhibition of apoptosis may worsen CF lung disease, supporting the concept that inhibition of apoptosis in the airways leads to delayed clearance of neutrophils and consequently to a hyperinflammatory state, and contributes to goblet cell metaplasia, representing a feature of CF airway disease.47,48 SNPs in the intergenic region of 11p13 may impair expression of EHF and APIP, but also that of other proximate genes such as ELF5 and PDHX, so studies are ongoing to elucidate the mechanism of disease pathogenesis resulting from genomic polymorphisms.44
Five genes (CBLN4, MC3R, CASS4, CSTF1, AURKA) were localized within the 20p13.2 linkage region, all of them being expressed in either fetal or adult lung or bronchial epithelial cells. CASS4, for instance, is a member of the CAS scaffolding protein family and plays a role in the tyrosine kinase-based signaling regulating cell adhesion and spreading.49 AURKA encodes an Aurora-related kinase A that is involved in various mitotic events, such as establishment of the mitotic spindle, centrosome maturation, chromosomal alignment, and cytokinesis, as well as in proper disassembly of primary cilia.50 The most interesting among the five genes, however, is MC3R, which has been implicated in regulation of energy balance and modulation of neutrophil accumulation in lung inflammation, both of which are key features of CF lung disease.51,52 Studies are underway to identify the mechanisms of lung disease that may reflect the linkage signal at 20p13.2.44
In addition, there were six other regions of suggestive association, among them the HLA class II DR locus on 6p21.3, which is well known to be involved in the manifestation of proinflammatory and autoimmune disorders, such as rheumatoid arthritis, type 1 diabetes, and multiple sclerosis, and the highly conserved ancestral haplotype 8.1 encoded in the HLA region has recently been reported to be associated with the severity of CF lung disease.53
In an exome-wide approach, Emond et al54 sequenced 43 CF patients with early onset of chronic Pseudomonas aeruginosa infection (≤5 years) as well as 48 individuals who had never had P. aeruginosa infection by age 14 years or beyond. Missense variants in a single gene, DCTN4 (dynactin 4), on chromosome 5q33.1 were found to be significantly associated with early age of chronic P. aeruginosa infection as well as of the first mucoid P. aeruginosa culture discovering DCTN4, a component of the dynein-dependent motor and part of the autophagy process, as an important genetic modifier of the CF phenotype. Although this strategy usually requires analysis of larger sample sizes, exome sequencing is an additional promising tool for identification of disease modifiers in cases of extreme phenotype study design (Tables 1 and 2).
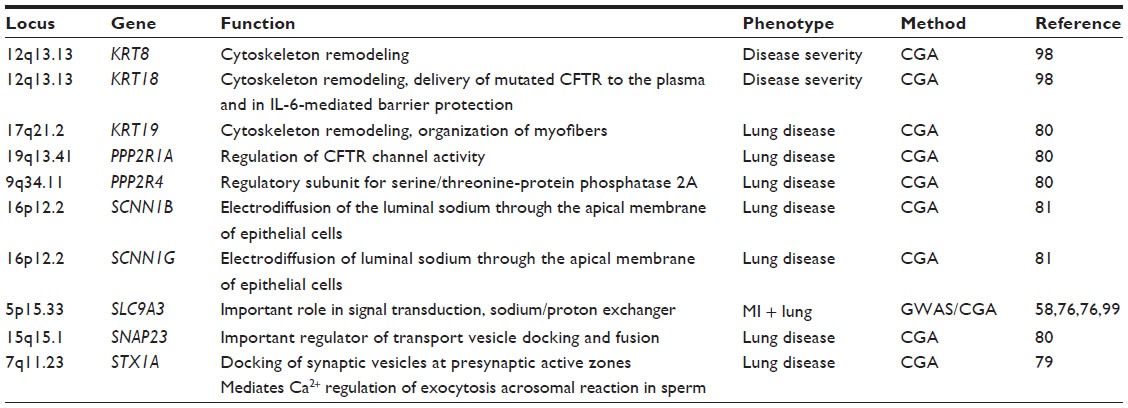 | Table 1 Direct modifiers of the CFTR interactome |
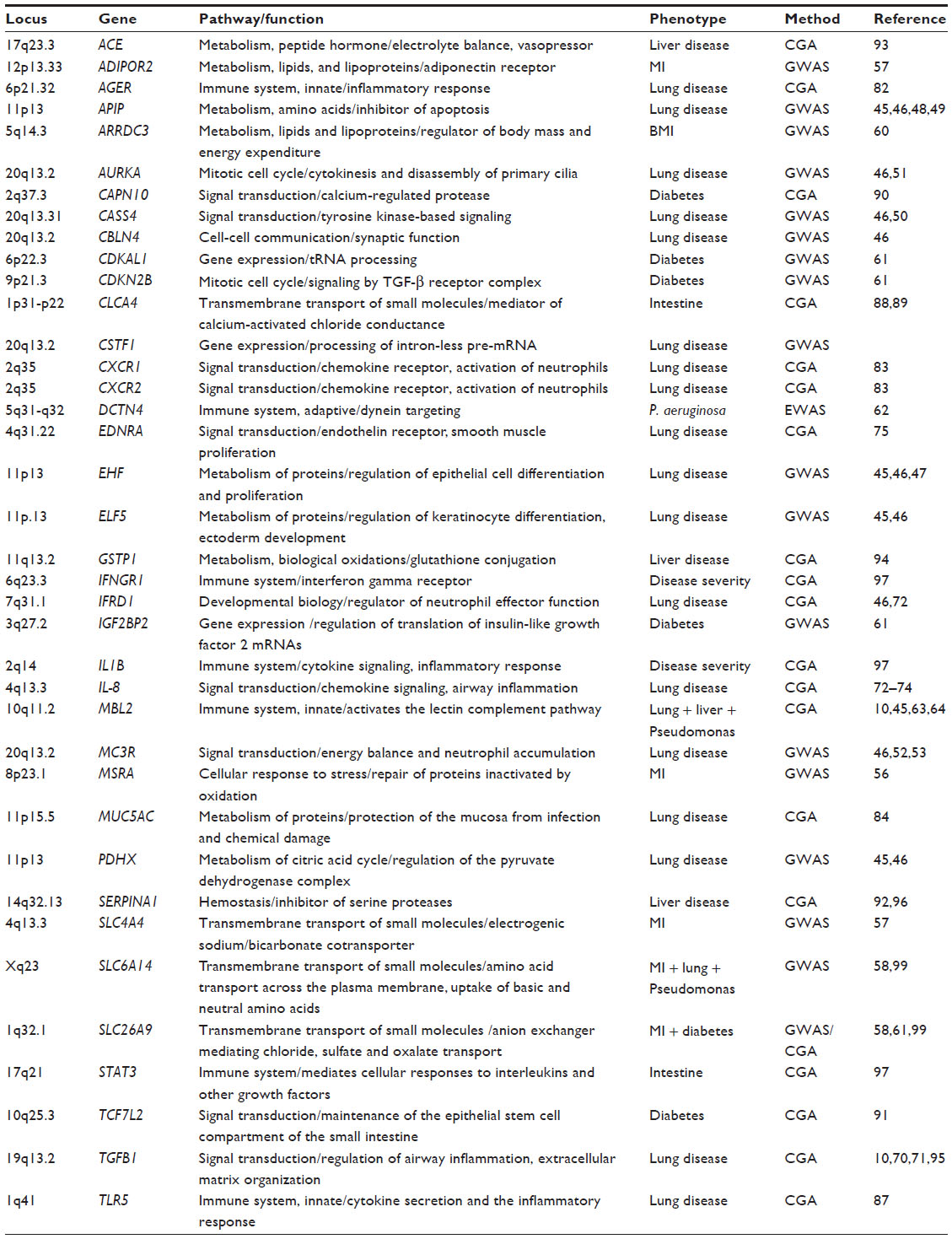 | Table 2 Indirect modifiers of different pathways |
CF in intestine cells
Using genome-wide analyses, Blackman et al55 identified several chromosomal regions suggesting contribution to (4q35.1, 8p23.1, 11q25) or protection from (20p11.22, 21q22.3) development of meconium ileus. By employing both linkage and transmission methods in a family-based study, followed by replication in an unrelated sample of CF patients, Henderson et al56 confirmed a strong association between a three-polymorphism haplotype corresponding to the MSRA gene on chromosome 8 and meconium ileus. Another linkage study replicated in an independent sample of CF families provided evidence for a locus promoting meconium ileus on chromosome 12p13.3 and pointed to the involvement of the adiponectin receptor 2 gene (ADIPOR2). Moreover, a protective locus was mapped to chromosome 4q13.3, including the sodium bicarbonate cotransporter SLC4A4 as a candidate gene.57 A conventional GWA study identified the association of several SNPs within two regions, including SLC26A9 on chromosome 1 (1q32.1) and SLC6A14 on chromosome X (Xq23–24), with meconium ileus in 3,763 affected individuals and these associations were replicated in an independent combined patient cohort from North America and France. A subsequent hypothesis-driven GWA study confirmed SLC26A9 and SLC6A14 and in addition SLC9A3 as modifiers of meconium ileus, but also provided evidence of a collective association of multiple constituents of the apical membrane with meconium ileus.58
In another approach, body mass index data collected from the CF Twin-Sibling Study (ClinicalTrials.Gov identifier NCT00037778)41 were analyzed to define the contribution of genetic modifiers to the nutritional status of young CF patients aged 5–10 years.59 For 1,010 individuals with pancreatic insufficiency, significant linkage of body mass index and the two chromosomal regions 1p36.1 and 5q14 was found, the latter including the arrestin domain-containing 3 (ARRDC3) gene, a regulator of body mass and energy expenditure in males.60
CF-related diabetes
A GWA study including 3,059 CF patients identified an association of SNPs within and 5′ to the SLC26A9 gene with CF-related diabetes. These findings were replicated in 694 individuals. In addition, SNPs for type 2 diabetes at or near CDKAL1, CDKN2A/B, and IGF2BP2 were found to be associated with CF-related diabetes as well.61 As SLC26A9 encodes an anion transporter conducting both chloride and bicarbonate, the authors speculate that “alteration in the expression of CFTR or SLC26A9 (or both) might affect β-cell function directly by altering chloride or bicarbonate flow (or both)”.
Candidate gene studies
CF lung disease
Defective ion transport resulting in disturbed mucociliary clearance and, as a consequence, in acute and chronic infections followed by an inflammatory response, form the basis of lung disease in CF. Therefore, genes involved in these pathways are expected to contribute to the CF phenotype by modifying the clinical course and severity of lung disease.
One of the first and most extensively studied modifier genes is MBL2, the gene that encodes mannose binding lectin (MBL) and that was selected as a candidate based on its critical role in the innate immune response and on the fact that MBL deficiency leads to a predilection for bacterial and viral infection. However, the findings of these numerous studies are in part contradictory, and the role of MBL2 as a genetic modifier of CF remains a matter of some debate. Six studies involving more than 2,000 CF patients demonstrated an association of MBL-deficient genotypes with more severe lung function, three studies reported earlier onset of P. aeruginosa infection, whereas several studies did not detect an association with lung disease and/or with age of infection with P. aeruginosa. In the most comprehensive study of MBL2 as a CF modifier, Dorfman et al62 evaluated the association of MBL2 genotypes with CF pulmonary phenotype in a cohort of 1,019 Canadian pediatric CF patients and demonstrated that MBL2 deficiency was significantly associated with an increased risk of early infection with P. aeruginosa and the rate of decline of pulmonary function in pediatric CF patients. Using a modeling framework considering confounding effects of related variables, such as lung disease severity, infection status, age, and CFTR genotype, McDougal et al63 showed that the MBL2 genotype was associated with infection status rather than with the other variables and that MBL deficiency primarily results in earlier P. aeruginosa infection and conversion to the mucoid form, leading presumably to more severe lung disease and reduced survival. Most of these studies have been reviewed and included in a meta-analysis by Chalmers et al,64 who concluded that the MBL2 gene is a modifier of CF lung disease and that MBL insufficiency is associated with earlier acquisition of P. aeruginosa, reduced lung function, and a higher rate of end-stage CF.
The gene second most often investigated for modifier effects is transforming growth factor beta 1 (TGFβ1), which is well known to be involved in the regulation of airway inflammation and remodeling65,66 and to influence the risk for airways disease in asthma67,68 and chronic obstructive pulmonary disease.69 The first large study reporting an association between TGFβ1 variants in the promoter (−509C>T) and exon 1 (codon 10, c.29C>T) and severity of CF lung disease in a cohort of 808 F508del homozygotes was published by Drumm et al.70 This finding was replicated in 498 patients with various genotypes and independently confirmed by Bremer et al,71 demonstrating association of a TGFβ1 haplotype (3′ C allele (rs8179181/-509C/c.29T) with improved lung function in CF. A few small studies reported no or not the same association between TGFβ1 and CF lung function.10
Gu et al72 identified the interferon-related developmental regulator 1 (IFRD1) gene as a modifier of the severity of CF lung disease. Via transcriptional mechanisms, IFRD1 regulates the neutrophil effector function during bacterial infection and may modulate the pathogenesis of CF lung disease through this pathway. Polymorphisms in IFRD1 showed association in a family-based (twin and siblings) population, but did not achieve genome-wide significance45 and have not been replicated in other CF populations.
Another gene, apart from MBL2, TGFβ1 and IFRD1, that is well known to be associated with airways inflammation in CF and suggested to modify lung disease severity by affecting host ability to tolerate infection,72 is interleukin-8 (IL-8). A modifier effect of variants in IL-8 was first noted in a CF cohort of 737 F508del homozygous patients and found to be most strongly pronounced in males.73 A second study in another 385 patients was able to confirm this finding for males;73 however, a further analysis of 329 patients failed to detect an association between lung disease and IL-8 variants.74
A well replicated association with CF lung disease has been described for the variant 6672G>C(rs5335) in the 3′ UTR of the endothelial receptor type A (EDNRA) gene. The C allele was more commonly found in CF patients with severe lung disease in four separate CF cohorts and correlated with increased EDNRA mRNA levels, resulting in increased smooth muscle proliferation and subsequent worsening of the CF pulmonary phenotype by altering smooth muscle tone in the airways and/or vasculature.75
As CFTR dysfunction leads to salt and water imbalance across airway epithelia and impaired mucociliary clearance, variants in the SLC9A3 gene encoding the sodium/hydrogen exchanger 3 (NHE3) were tested and found to be associated with susceptibility to bacterial infections and severity of CF lung disease.76 SLC9A3 reciprocally interacts with CFTR through a regulatory complex including NHERF-2, ezrin, and protein kinase A, and therefore provides plausible genetic evidence of a modulatory effect.77 Additional genes of the CFTR interactome, such as SCNN1B and SCNN1G encoding the β-subunits and γ-subunits of ENaC,78 STX1A,79 KRT19, PPP2R4, PPP2R1A, and SNAP23,80 have been found to be involved in the modification of CF lung disease. While the modulating effect of SCNN1B and SCNN1G could not be confirmed,81 the influence of STX1A on lung disease severity was replicated in an independent CF patient cohort and variant rs2228607 was shown to reinforce aberrant splicing of STX1A mRNA, triggering nonsense-mediated mRNA decay and indicating functional relevance of this variant.79 The findings by Gisler et al80 are based upon longitudinal data for six different lung function parameters and are waiting replication; however, to have comparable longitudinal data from an independent CF patient cohort at hand is not simple.
Further associations with CF lung disease have been described, but not yet replicated, for the variant −429T/C in the promoter of the AGER gene, for SNPs in CXCR1 and CXCR2, and for a 6.4 kb VNTR allele of the secreted mucin gene MUC5AC. The AGER allele −429C has been shown to lead to an increased promoter activity upregulating RAGE, that triggers the generation of reactive oxygen species and activation of signal transduction pathways, and excessive expression of which may result in increased airway inflammation and enhanced lung disease severity.82 The chemokine receptors CXCR1 and CXCR2 play an important role in neutrophilic inflammation, and a specific CXCR1/2 haplotype cluster has been identified that is associated with lung function in CF patients and is thought to modulate the pulmonary outcome through a dysregulation of neutrophilic effector functions.83 As the MUC5AC gene may be involved in mucociliary clearance, Guo et al84 speculate that the 6.4 kb severity-associated VNTR allele could alter the function of the airway mucus, affecting the inflammation status and progression of lung disease.
Clinical trials have demonstrated that anti-inflammatory therapy may improve lung function and body weight in CF patients,85 and emerging evidence indicates that inhibition of the flagellin receptor TLR5 normalizes the inflammatory response generated by CF airway epithelial cells after exposure to P. aeruginosa.86 To validate TLR5 as a disease modifier and anti-inflammatory target, Blohmke et al87 examined the relationship between the TLR5SNP c.1174C>T (rs5744168) leading to a premature stop codon and the two clinical parameters of lung function and body weight in 2,219 CF patients. They were able to demonstrate that the TLR5 allele c.1174T significantly decreases TLR5 responsiveness and associates with improved nutritional status in adults with CF, providing evidence that therapeutic strategies to inhibit TLR5 may be beneficial for the clinical outcome of CF patients (Tables 1 and 2).
CF in intestinal cells
The predisposition to meconium ileus in CF is clearly genetically determined; however, the candidate gene approach has not been applied to this CF trait so far. The CLCA gene locus has previously been identified as a modulator of the basic gastrointestinal defect in CF.88 Using a customized SNP map with a total of 17 diallelic markers and a resequencing strategy for fine-mapping, Kolbe et al89 detected significant differences in allele distribution for markers within the promoter of the CLCA4 gene among F508del homozygous patients with either no, CFTR-mediated, or Ca2+-mediated residual chloride conductance. These findings suggest that CLCA4 modulates the capability to express residual chloride secretion in colonic tissue and therefore acts as a modifier of the basic defect in intestinal cells of CF patients.
CF-related diabetes
The fact that diabetes is a common complication of CF and strongly influenced by modifier genes and that a family history of type 2 diabetes increases the risk for CF-related diabetes raises the need to test T2DM-associated genes as potential modifiers of CF-related diabetes. Analyzing six genes contributing to the manifestation of type 2 diabetes, Derbel et al90 presented the first evidence for an involvement of a polymorphism in the CAPN10 gene in the pathogenesis of CF-related diabetes. A further candidate gene approach revealed the modifying effects of a variant in the TCF7L2 gene91 that associates with both type 2 diabetes and CF-related diabetes, supporting the concept that diabetes develops in patients who may have underlying susceptibility to β-cell dysfunction.
CF-related liver disease
Only a subset (3%–5%) of CF patients develops severe liver disease (CFLD), characterized by cirrhosis with portal hypertension indicating contribution of non-CFTR genetic variability to the risk for CFLD. The largest two-stage study focused on nine functional variants in five candidate genes (SERPINA1, ACE, GSTP1, MBL2, TGFβ1) previously studied in CFLD, but including only small and heterogeneous patient cohorts with and without portal hypertension.92–95 Two large populations of CF patients with (128/971 and 139/1,227, respectively) and without (843/971 and 1,088/1,227, respectively) liver disease and portal hypertension were tested, and only the SERPINA1 Z-allele was found to significantly associate with CFLD and portal hypertension.96 TGFβ1 variants showed association with CFLD in stage 1, but did not replicate in stage 2 of the study.
Manifestation and severity of CF disease
An association study comparing F508del homozygous CF patient subsets selected for an extreme clinical phenotype and/or their basic defect manifestation tested the genes STAT3, IL1B, and IFNGR1 as modifiers of CF disease. Evidence for allelic association was found for STAT3 and CFTR-mediated residual chloride secretion in intestinal tissue, for IL1B and disease severity, and for IFNGR1 and intrapair discordance, implying that immunorelevant pathways and ion secretion merge at the level of epithelial cells to integrate the signaling of cytokines due to innate and acquired immune defense.97
Another candidate gene approach investigated the two neighboring cytokeratin genes KRT8 and KRT18 in F508del homozygous sibling pairs and unrelated index cases selected for their extreme clinical or electrophysiological phenotype characterized by intestinal current or nasal potential difference measurements. KRT8, but not KRT18, showed association with CF disease severity and CFTR-mediated residual chloride secretion. Absence of chloride secretion was determined by the recessive haplotype 1122 (rs1907671, rs4300473, rs2035878, rs2035875), whereas the contrasting haplotype 2211 was dominant for the presence of CFTR-mediated residual chloride secretion and associated with a mild phenotype. The significance of these results for KRT8 is that modifiers of CF disease severity can be recognized through their association with the basic defect, which is less influenced by environmental factors.98
Li et al99 tested the hypothesis that the same risk alleles for meconium ileus in SLC26A9, SLC9A3, and SLC6A14 are pleiotropic for severity of lung disease, age at first P. aeruginosa infection, and early exocrine pancreatic disease. They found that SLC26A9 was pleiotropic for meconium ileus and pancreatic damage at rs7512462, SLC9A3 for meconium ileus and lung disease at rs17563161, and SLC6A14 for meconium ileus and both lung disease and age at first P. aeruginosa infection at rs3788766. These findings demonstrate that pleiotropy in a multiorgan disease has to be checked for and can be assessed in a single study sample. Evidence of pleiotropy for (modifier) genes may enable the development of complementary therapeutic strategies that will benefit multiple disease-associated tissues.
Implications for other monogenic disorders
The impact of disease-modifying genes is manifold because understanding the diverse genetic contribution to a disease phenotype has the potential to improve prognosis, to develop new therapeutic approaches, and to open the door to personalized medicine. The experience in CF modifier research represents a useful basis for other single gene disorders mainly regarding consistent phenotype measures and study designs. To identify modifying loci and genes and to replicate associations, both genome-wide methods and candidate gene approaches are needed, including large sample sizes and phenotypically well defined homogeneous patient cohorts, respectively. Further, the role of genetic factors in the variability of a disease has to be evidenced by family studies. Genotyping of parents allows reliable assessment of descent and detection of transmission disequilibrium, whereas twin and sibling analyses are crucial to prove or exclude phenotypic correlation within families and to define the genetic model underlying variability in disease expression.
It has to be considered that modifier genes may each contribute small effects to a disease phenotype, but may work in concert with environmental influences, resulting in an amplified or diminished consequence. Thus, longitudinal studies of patients with monogenic disorders are needed to define key factors contributing to gene–environment interactions. Moreover, the admixture of ethnic groups plays an important role in the characterization of modifying effects, as modifier genes are diverse in different ethnicities and their complexity increases in ethnically admixed populations according to the extent of gene flow.2
The increase in modifier gene identification as a consequence of new genetic technologies leads to the conclusion that epistasis may represent a principal mechanism explaining the individual phenotypic variability of most genetic diseases.
Diagnostic and therapeutic significance
Currently, it is very difficult to decide if a CFTR mutation is disease-causing or rather a neutral variant and if a specific gene is modifying the CF phenotype or not. Thus, correlation of genotype and basic defect phenotype (CFTR protein) remains an essential approach to directly measure the influence of CFTR mutations on subsequent clinical manifestations. However, identification of risk alleles in modifier genes and understanding their contribution to phenotypic variation may open the possibility of defining risk factors early in life, allowing more accurate diagnosis in due time and providing better prediction of disease course. With the advent of next-generation sequencing, large numbers of genes and genetic variants can easily be detected, enabling individual genotyping of primary disease genes and modifier genes in one single step and within a short time. Assuming correct understanding and interpretation of these data, complex genetic testing including the entire panel of variants known to contribute to the phenotype of a specific disease will significantly improve diagnosis for any monogenic disorder. Moreover, such a diagnostic approach may also facilitate personalized treatment according to a given patient’s genetic profile. For example, CF patients with a modifier allele constellation predicting early P. aeruginosa infection and conversion to the mucoid form may benefit from earlier and more rigorous antibiotic applications than those commonly used.
The identification of genes that influence clinical phenotypes opens a new field of therapeutic approaches in that not only proteins of primary disease-causing genes but also of disease modifiers can be targeted to potentiate residual activities of the primary gene products or to fully compensate for their defective function. However, it has to be taken into account that effects of modifiers of CF and other monogenic disorders may be limited to an individual disease phase, age, sex, or a specific organ or tissue. Using CF again as an example, anti-inflammatory therapy may not have the expected effect in young patients due to the unwanted suppression of immune responses to sporadic infections, while the same therapy could improve the health of older patients with established chronic infections.100 Therefore, strategies for the development of new drugs and therapies via modifiers should focus on specific, well defined patient groups (eg, specific age, disease severity, genotype) that are likely to benefit most from the therapeutic application. Moreover, the sequential application of different drugs according to, eg, age, disease stage, and/or a combination therapy (several drugs at the same time) may result in the highest response rate and best improvement in health.
Conclusion
Key reasons for identifying disease modifiers are to improve prognosis of disease course and outcome and to aid the development of therapies. Even though the primary defect in CF is monogenic by definition, the manifestation of endophenotypes such as diabetes or liver disease as well as disease severity are rather polygenic by nature, meaning that searching for modifiers in CF or any other monogenic disorder raises the same hurdles as characterization of other complex diseases, such as asthma or cardiovascular disorders. Nevertheless, the sum of modifier genes influencing the CF phenotype that has been reported so far is impressive and provides novel insight into biological pathways and disease pathophysiology, paving the way to the understanding of other genetic disorders. Exome-wide and genome-wide analyses will become routine in the near future and may improve diagnosis and phenotype prediction of genetic disorders by including all genes known to be involved in the manifestation of a specific disease. In addition, proteins encoded by genes that have been proven to modify the disease course will become promising targets for therapeutic interventions.
In summary, modifier research opens new diagnostic, prognostic, and therapeutic perspectives, leading at the same time to the understanding that “there is no such thing as a “single” disorder” and that “genetic diseases represent a continuum with diminishing influence from a single primary gene influenced by modifier genes, to increasingly shared influence by multiple genes”,101 making genetics even more complex and complicated than before.
Disclosure
The author reports no conflicts of interests in this work.
References
Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11(20):2463–2468. | |
Nagel RL. Epistasis and the genetics of human diseases. C R Biol. 2005;328(7):606–615. | |
Gropman AL, Adams DR. Atypical patterns of inheritance. Semin Pediatr Neurol. 2007;14(1):34–45. | |
Slavotinek A, Biesecker LG. Genetic modifiers in human development and malformation syndromes, including chaperone proteins. Hum Mol Genet. 2003;12 Spec No 1:R45–R50. | |
Genin E, Feingold J, Clerget-Darpoux F. Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet. 2008;124(4):357–368. | |
Rosenstein BJ, Zeitlin PL. Cystic fibrosis. Lancet. 1998;351(9098):277–282. | |
Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med. 2002;347(6):439–442. | |
Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173(5):475–482. | |
Kerem E, Corey M, Kerem B, Durie P, Tsui LC, Levison H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr. 1989;114(5):767–773. | |
Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214:57–69. | |
Ooi CY, Dorfman R, Cipolli M, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. 2011;140(1):153–161. | |
Watelet JB, Van Cauwenberge P, Bachert C. Rhinological aspects of cystic fibrosis. Monaldi Arch Chest Dis. 2000;55(6):475–477. | |
Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10 Suppl 2:S86–S102. | |
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. | |
Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–1065. | |
Tsui LC, Dorfman R. The cystic fibrosis gene: a molecular genetic perspective. Cold Spring Harb Perspect Med. 2013;3(2):a009472. | |
Bradbury NA. Intracellular CFTR: localization and function. Physiol Rev. 1999;79(Suppl 1):S175–S191. | |
Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003;285(1):C1–C18. | |
Kim D, Steward MC. The role of CFTR in bicarbonate secretion by pancreatic duct and airway epithelia. J Med Invest. 2009;56 Suppl:336–342. | |
Quinton PM. Role of epithelial HCO3(-) transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol. 2010;299(6):C1222–C1233. | |
Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. | |
Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. | |
Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. | |
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. | |
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. | |
MacDonald KD, McKenzie KR, Zeitlin PL. Cystic fibrosis transmembrane regulator protein mutations: ‘class’ opportunity for novel drug innovation. Paediatr Drugs. 2007;9(1):1–10. | |
Haardt M, Benharouga M, Lechardeur D, Kartner N, Lukacs GL. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J Biol Chem. 1999;274(31):21873–21877. | |
Ferrari M, Cremonesi L. Genotype-phenotype correlation in cystic fibrosis patients. Ann Biol Clin (Paris). 1996;54(6):235–241. | |
Gallati S. Genetics of cystic fibrosis. Semin Respir Crit Care Med. 2003;24(6):629–638. | |
Kerem E, Corey M, Kerem BS, et al. The relation between genotype and phenotype in cystic fibrosis – analysis of the most common mutation (delta F508). N Engl J Med. 1990;323(22):1517–1522. | |
Borgo G, Gasparini P, Bonizzato A, Cabrini G, Mastella G, Pignatti PF. Cystic fibrosis: the delta F508 mutation does not lead to an exceptionally severe phenotype. A cohort study. Eur J Pediatr. 1993;152(12):1006–1011. | |
Hubert D, Bienvenu T, Desmazes-Dufeu N, et al. Genotype-phenotype relationships in a cohort of adult cystic fibrosis patients. Eur Respir J. 1996;9(11):2207–2214. | |
Salvatore F, Scudiero O, Castaldo G. Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am J Med Genet. 2002;111(1):88–95. | |
Kraemer R, Birrer P, Liechti-Gallati S. Genotype-phenotype association in infants with cystic fibrosis at the time of diagnosis. Pediatr Res. 1998;44(6):920–926. | |
Kraemer R, Aebi C, Casaulta Aebischer C, Gallati S. Early detection of lung disease and its association with the nutritional status, genetic background and life events in patients with cystic fibrosis. Respiration. 2000;67(5):477–490. | |
Kraemer R, Latzin P, Pramana I, Ballinari P, Gallati S, Frey U. Long-term gas exchange characteristics as markers of deterioration in patients with cystic fibrosis. Respir Res. 2009;10:106. | |
Highsmith WE, Burch LH, Zhou Z, et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J Med. 1994;331(15):974–980. | |
Mak V, Jarvi KA, Zielenski J, Durie P, Tsui LC. Higher proportion of intact exon 9 CFTR mRNA in nasal epithelium compared with vas deferens. Hum Mol Genet. 1997;6(12):2099–2107. | |
Mekus F, Ballmann M, Bronsveld I, Bijman J, Veeze H, Tummler B. Categories of deltaF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res. 2000;3(4):277–293. | |
Mekus F, Laabs U, Veeze H, Tummler B. Genes in the vicinity of CFTR modulate the cystic fibrosis phenotype in highly concordant or discordant F508del homozygous sib pairs. Hum Genet. 2003;112(1):1–11. | |
Vanscoy LL, Blackman SM, Collaco JM, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med. 2007;175(10):1036–1043. | |
Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther. 2005;108(2):208–223. | |
Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7(6):426–436. | |
Knowles MR, Drumm M. The influence of genetics on cystic fibrosis phenotypes. Cold Spring Harb Perspect Med. 2012;2(12):a009548. | |
Wright FA, Strug LJ, Doshi VK, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 2011;43(6):539–546. | |
Stanke F, van Barneveld A, Hedtfeld S, Wolfl S, Becker T, Tummler B. The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur J Hum Genet. 2014;22(5):660–666. | |
Dibbert B, Weber M, Nikolaizik WH, et al. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci U S A. 1999;96(23):13330–13335. | |
Harris JF, Fischer MJ, Hotchkiss JR, et al. Bcl-2 sustains increased mucous and epithelial cell numbers in metaplastic airway epithelium. Am J Respir Crit Care Med. 2005;171(7):764–772. | |
Tikhmyanova N, Tulin AV, Roegiers F, Golemis EA. Dcas supports cell polarization and cell-cell adhesion complexes in development. PLoS One. 2010;5(8):e12369. | |
Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–1363. | |
Savastano DM, Tanofsky-Kraff M, Han JC, et al. Energy intake and energy expenditure among children with polymorphisms of the melanocortin-3 receptor. Am J Clin Nutr. 2009;90(4):912–920. | |
Getting SJ, Riffo-Vasquez Y, Pitchford S, et al. A role for MC3R in modulating lung inflammation. Pulm Pharmacol Ther. 2008;21(6):866–873. | |
Corvol H, Beucher J, Boelle PY, et al. Ancestral haplotype 8.1 and lung disease severity in European cystic fibrosis patients. J Cyst Fibros. 2012;11(1):63–67. | |
Emond MJ, Louie T, Emerson J, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44(8):886–889. | |
Blackman SM, Deering-Brose R, McWilliams R, et al. Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology. 2006;131(4):1030–1039. | |
Henderson LB, Doshi VK, Blackman SM, et al. Variation in MSRA modifies risk of neonatal intestinal obstruction in cystic fibrosis. PLoS Genet. 2012;8(3):e1002580. | |
Dorfman R, Li W, Sun L, et al. Modifier gene study of meconium ileus in cystic fibrosis: statistical considerations and gene mapping results. Hum Genet. 2009;126(6):763–778. | |
Sun L, Rommens JM, Corvol H, et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet. 2012;44(5):562–569. | |
Bradley GM, Blackman SM, Watson CP, Doshi VK, Cutting GR. Genetic modifiers of nutritional status in cystic fibrosis. Am J Clin Nutr. 2012;96(6):1299–1308. | |
Patwari P, Emilsson V, Schadt EE, et al. The arrestin domain-containing 3 protein regulates body mass and energy expenditure. Cell Metab. 2011;14(5):671–683. | |
Blackman SM, Commander CW, Watson C, et al. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes. 2013;62(10):3627–3635. | |
Dorfman R, Sandford A, Taylor C, et al. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest. 2008;118(3):1040–1049. | |
McDougal KE, Green DM, Vanscoy LL, et al. Use of a modeling framework to evaluate the effect of a modifier gene (MBL2) on variation in cystic fibrosis. Eur J Hum Genet. 2010;18(6):680–684. | |
Chalmers JD, Fleming GB, Hill AT, Kilpatrick DC. Impact of mannose-binding lectin insufficiency on the course of cystic fibrosis: a review and meta-analysis. Glycobiology. 2011;21(3):271–282. | |
Akhurst RJ. TGF beta signaling in health and disease. Nat Genet. 2004;36(8):790–792. | |
Yang YC, Zhang N, Van Crombruggen K, Hu GH, Hong SL, Bachert C. Transforming growth factor-beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy. 2012;67(10):1193–1202. | |
Silverman ES, Palmer LJ, Subramaniam V, et al. Transforming growth factor-beta1 promoter polymorphism C-509T is associated with asthma. Am J Respir Crit Care Med. 2004;169(2):214–219. | |
Chiang CH, Chuang CH, Liu SL, Shen HD. Genetic polymorphism of transforming growth factor beta1 and tumor necrosis factor alpha is associated with asthma and modulates the severity of asthma. Respir Care. 2013;58(8):1343–1350. | |
Wu L, Chau J, Young RP, et al. Transforming growth factor-beta1 genotype and susceptibility to chronic obstructive pulmonary disease. Thorax. 2004;59(2):126–129. | |
Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353(14):1443–1453. | |
Bremer LA, Blackman SM, Vanscoy LL, et al. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet. 2008;17(14):2228–2237. | |
Gu Y, Harley IT, Henderson LB, et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature. 2009; 458(7241):1039–1042. | |
Hillian AD, Londono D, Dunn JM, et al. Modulation of cystic fibrosis lung disease by variants in interleukin-8. Genes Immun. 2008;9(6):501–508. | |
Corvol H, Boelle PY, Brouard J, et al. Genetic variations in inflammatory mediators influence lung disease progression in cystic fibrosis. Pediatr Pulmonol. 2008;43(12):1224–1232. | |
Darrah R, McKone E, O’Connor C, et al. EDNRA variants associate with smooth muscle mRNA levels, cell proliferation rates, and cystic fibrosis pulmonary disease severity. Physiol Genomics. 2010;41(1):71–77. | |
Dorfman R, Taylor C, Lin F, et al. Modulatory effect of the SLC9A3 gene on susceptibility to infections and pulmonary function in children with cystic fibrosis. Pediatr Pulmonol. 2011;46(4):385–392. | |
Favia M, Fanelli T, Bagorda A, et al. NHE3 inhibits PKA-dependent functional expression of CFTR by NHERF2 PDZ interactions. Biochem Biophys Res Commun. 2006;347(2):452–459. | |
Stanke F, Becker T, Cuppens H, et al. The TNFalpha receptor TNFRSF1A and genes encoding the amiloride-sensitive sodium channel ENaC as modulators in cystic fibrosis. Hum Genet. 2006;119(3):331–343. | |
von Kanel T, Stanke F, Weber M, et al. Clinical and molecular characterization of the potential CF disease modifier syntaxin 1A. Eur J Hum Genet. 2013;21(12):1462–1466. | |
Gisler FM, von Kanel T, Kraemer R, Schaller A, Gallati S. Identification of SNPs in the cystic fibrosis interactome influencing pulmonary progression in cystic fibrosis. Eur J Hum Genet. 2013;21(4):397–403. | |
Viel M, Leroy C, Hubert D, Fajac I, Bienvenu T. ENaCbeta and gamma genes as modifier genes in cystic fibrosis. J Cyst Fibros. 2008;7(1):23–29. | |
Beucher J, Boelle PY, Busson PF, Muselet-Charlier C, Clement A, Corvol H. AGER -429T/C is associated with an increased lung disease severity in cystic fibrosis. PLoS One. 2012;7(7):e41913. | |
Kormann MS, Hector A, Marcos V, et al. CXCR1 and CXCR2 haplotypes synergistically modulate cystic fibrosis lung disease. Eur Respir J. 2012;39(6):1385–1390. | |
Guo X, Pace RG, Stonebraker JR, et al. Mucin variable number tandem repeat polymorphisms and severity of cystic fibrosis lung disease: significant association with MUC5AC. PLoS One. 2011;6(10):e25452. | |
Lai HC, Kosorok MR, Laxova A, Davis LA, FitzSimmon SC, Farrell PM. Nutritional status of patients with cystic fibrosis with meconium ileus: a comparison with patients without meconium ileus and diagnosed early through neonatal screening. Pediatrics. 2000;105(1 Pt 1):53–61. | |
Blohmke CJ, Victor RE, Hirschfeld AF, et al. Innate immunity mediated by TLR5 as a novel antiinflammatory target for cystic fibrosis lung disease. J Immunol. 2008;180(11):7764–7773. | |
Blohmke CJ, Park J, Hirschfeld AF, et al. TLR5 as an anti-inflammatory target and modifier gene in cystic fibrosis. J Immunol. 2010;185(12):7731–7738. | |
Ritzka M, Stanke F, Jansen S, et al. The CLCA gene locus as a modulator of the gastrointestinal basic defect in cystic fibrosis. Hum Genet. 2004;115(6):483–491. | |
Kolbe EW, Tamm S, Hedtfeld S, Becker T, Tummler B, Stanke F. CLCA4 variants determine the manifestation of the cystic fibrosis basic defect in the intestine. Eur J Hum Genet. 2013;21(6):691–694. | |
Derbel S, Doumaguet C, Hubert D, et al. Calpain 10 and development of diabetes mellitus in cystic fibrosis. J Cyst Fibros. 2006;5(1):47–51. | |
Blackman SM, Hsu S, Ritter SE, et al. A susceptibility gene for type 2 diabetes confers substantial risk for diabetes complicating cystic fibrosis. Diabetologia. 2009;52(9):1858–1865. | |
Frangolias DD, Ruan J, Wilcox PJ, et al. Alpha 1-antitrypsin deficiency alleles in cystic fibrosis lung disease. Am J Respir Cell Mol Biol. 2003;29(3 Pt 1):390–396. | |
Arkwright PD, Pravica V, Geraghty PJ, et al. End-organ dysfunction in cystic fibrosis: association with angiotensin I converting enzyme and cytokine gene polymorphisms. Am J Respir Crit Care Med. 2003;167(3):384–389. | |
Henrion-Caude A, Flamant C, Roussey M, et al. Liver disease in pediatric patients with cystic fibrosis is associated with glutathione S-transferase P1 polymorphism. Hepatology. 2002;36(4 Pt 1):913–917. | |
Gabolde M, Hubert D, Guilloud-Bataille M, Lenaerts C, Feingold J, Besmond C. The mannose binding lectin gene influences the severity of chronic liver disease in cystic fibrosis. J Med Genet. 2001;38(5):310–311. | |
Bartlett JR, Friedman KJ, Ling SC, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302(10):1076–1083. | |
Labenski H, Hedtfeld S, Becker T, Tummler B, Stanke F. Initial interrogation, confirmation and fine mapping of modifying genes: STAT3, IL1B and IFNGR1 determine cystic fibrosis disease manifestation. Eur J Hum Genet. 2011;19(12):1281–1288. | |
Stanke F, Hedtfeld S, Becker T, Tummler B. An association study on contrasting cystic fibrosis endophenotypes recognizes KRT8 but not KRT18 as a modifier of cystic fibrosis disease severity and CFTR mediated residual chloride secretion. BMC Med Genet. 2011;12:62. | |
Li W, Soave D, Miller MR, et al. Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum Genet. 2014;133(2):151–161. | |
Dorfman R. Modifier gene studies to identify new therapeutic targets in cystic fibrosis. Curr Pharm Des. 2012;18(5):674–682. | |
Dipple KM, McCabe ER. Modifier genes convert “simple” Mendelian disorders to complex traits. Mol Genet Metab. 2000;71(1–2):43–50. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.