")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Diagnosis and activity assessment of immunoglobulin A nephropathy: current perspectives on noninvasive testing with aberrantly glycosylated immunoglobulin A-related biomarkers
Authors Suzuki Y, Suzuki H, Makita Y, Takahata A, Takahashi K, Muto M, Sasaki Y, Kelimu A, Matsuzaki K, Yanagawa H, Okazaki K, Tomino Y
Received 12 August 2014
Accepted for publication 18 September 2014
Published 30 October 2014 Volume 2014:7 Pages 409—414
DOI https://doi.org/10.2147/IJNRD.S50513
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Pravin Singhal
Yusuke Suzuki,1 Hitoshi Suzuki,1 Yuko Makita,1 Akiko Takahata,1 Keiko Takahashi,1 Masahiro Muto,1 Yohei Sasaki,1 Atikemu Kelimu,1 Keiichi Matsuzaki,2 Hiroyuki Yanagawa,1 Keiko Okazaki,1 Yasuhiko Tomino1
1Division of Nephrology, Department of Internal Medicine, Juntendo University Faculty of Medicine, Tokyo, 2Kyoto University Health Service, Kyoto, Japan
Abstract: Immunoglobulin (Ig) A nephropathy (IgAN) is the most common form of glomerular disease worldwide and is associated with a poor prognosis. Thus, development of a curative treatment and strategies for early diagnosis and treatment are urgently needed. Pathological analysis of renal biopsy is the gold standard for the diagnosis and assessment of disease activity; however, immediate and frequent assessment based on biopsy specimens is difficult. Therefore, a simple and safe alternative is desirable. On the other hand, it is now widely accepted that multi-hit steps, including production of aberrantly glycosylated serum IgA1 (first hit), and IgG or IgA autoantibodies that recognize glycan containing epitopes on glycosylated serum IgA1 (second hit) and their subsequent immune complex formation (third hit) and glomerular deposition (fourth hit), are required for continued progression of IgAN. Although the prognostic and predictive values of several markers have been discussed elsewhere, we recently developed a highly sensitive and specific diagnostic method by measuring serum levels of glycosylated serum IgA1 and related IgA immune complex. In addition, we confirmed a significant correlation between serum levels of these essential effector molecules and disease activity after treatment, suggesting that each can be considered as a practical surrogate marker of therapeutic effects in this slowly progressive disease. Such a noninvasive diagnostic and activity assessment method using these disease-oriented specific biomarkers may be useful in the early diagnosis of and intervention in IgAN, with appropriate indication for treatment, and thus aid in the future development and dissemination of specific and curative treatments.
Keywords: galactose-deficient immunoglobulin A1, anti-glycan antibody, immune complex, N-acetylgalactosamine, surrogate marker
Limitations of renal biopsy and urinalysis in assessment of disease activity
Immunoglobulin A nephropathy (IgAN) is the most common form of primary glomerulonephritis worldwide, and is associated with a poor prognosis, resulting in end-stage kidney disease in approximately 40% of cases.1–3 Because the poor prognosis of IgAN is partly a result of delayed diagnosis, strategies for early diagnosis leading to early and effective medical intervention are urgently needed.
Pathological analysis of renal biopsy tissue is the gold standard for diagnosis of IgAN as well as assessment of disease activity and renal prognosis. However, the findings may differ according to the timing of renal biopsy during the 20-year course of IgAN.1,2 Different intervention periods may yield different findings on renal biopsy. For example, in studies using a renal biopsy database from the southern USA, IgAN was commonly identified in renal biopsy tissue specimens from patients aged <40 years, whereas focal glomerulosclerosis was reportedly the primary finding in patients aged ≥40 years.4 The authors pointed out that although primary focal glomerulosclerosis was most common in non-white individuals aged ≥40 years, secondary focal glomerulosclerosis was more likely in Caucasians because of “burned-out” IgAN. Moreover, renal biopsy is not frequently performed because of procedural risks and/or limitation of insurance coverage. These observations emphasize that renal biopsy provides only a snap-shot of the disease status and thus also has limitations in the assessment of disease activity. Furthermore, even if biopsy is performed early in the course of disease, findings may be inconclusive and prognosis may be difficult to predict. Even mild IgAN, presenting as hematuria or mild proteinuria with mild histological lesions at the time of renal biopsy, progresses to renal failure in 30% of cases.3,5,6 To determine disease stage, a noninvasive, real-time activity assessment method separate from renal biopsy is desirable.
Urinalysis is often used to assess disease activity, although this method also has limitations and restrictions. However, to date, there is no clear method to distinguish proteinuria from acute glomerular inflammatory lesions such as cellular crescents or burned-out sclerotic glomerulus in IgAN. Therefore, the rationale for treatment indication based on urinary protein levels in many clinical guidelines must be rigorously limited.
Taken together, activity assessment methods other than renal biopsy and urinalysis are needed. In addition to an objective method to assess disease activity, a simple and safe method of early diagnosis using valid biomarkers based on the pathogenesis of IgAN should be established.
Aberrantly glycosylated IgA and related IgA immune complexes as effector molecules in IgAN
Levels of the polymeric form of IgA are elevated in the serum of IgAN patients, whereas glomerular IgA in this disease is mainly of the IgA1 subtype.7,8 IgA1 has a longer hinge region, which can be glycosylated with various O-linked-glycans with N-acetylgalactosamine (GalNAc), galactose, and sialic acid, depending on the activity of specific glycosyltransferases.9 Previous reports have indicated that glomerular IgA1 in IgAN is either under-galactosylated.10,11 Therefore, aberrantly glycosylated IgA1 has long been considered as a possible cause of IgAN.12 In recent years, an increase in serum galactose-deficient IgA1 levels in patients with IgAN was quantitatively confirmed for the first time by Moldoveanu et al by use of an enzyme-linked immunosorbent assay using helix aspersa agglutinin lectin, which recognizes GalNAc residues.13 Moreover, Suzuki et al established immortalized B-cell lines that produce IgA1 from peripheral blood B cells obtained from IgAN patients and analyzed the characteristics of GdIgA1.14 IgA1 from IgAN was polymeric-dominant and galactose-deficient or over-sialylated on terminal GalNAc.9,13 In addition, enzyme analysis revealed abnormal enzymatic activity, such as a decrease in β1,3-galactosyltransferase and an increase in α2,6-sialyltransferase activities, which are critical for incorporation of galactose and sialic acid into GalNAc, respectively.14 Recent studies have shown that these enzymatic activities are possibly regulated by genetic mechanisms and dysregulation of mucosal immunity.15–17
A question arises regarding where such GdIgA1 is generated. Some clinical evidence, such as exacerbation of IgAN after upper respiratory tract infection, suggests mucosal contribution in the pathogenesis of IgAN. Indeed, some Japanese groups demonstrated aberrant glycosylation in IgA from palatine tonsils of IgAN patients,18,19 whereas some Chinese groups reported abnormal cytokine profiles in the tonsils leading to an imbalance in β1,3-galactosyltransferase and an increase in α2,6-sialyltransferase,20,21 indicating that palatine tonsils may be the primary site of GdIgA1.
Our recent studies further confirmed this notion. Most IgAN patients have decreased serum IgA levels after tonsillectomy. In our analysis, we found an average decrease of nearly 10% at 4 weeks after tonsillectomy. Moreover, patients demonstrating a relatively large decrease in serum IgA levels (>10%) achieved better clinical outcomes after tonsillectomy,22 suggesting that nephritogenic IgA may play a role in the decrease in IgA after tonsillectomy. Indeed, high serum IgA levels (>315 mg/dL) and serum IgA/C3 ratio (>3.01) in association with hematuria (more than five red blood cells per high power field) and proteinuria (>0.3 g/day) have a diagnostic value for IgAN.23 To further confirm this idea, we directly evaluated changes in serum GdIgA1 levels by helix aspersa agglutinin lectin enzyme-linked immunosorbent assay before and 4 weeks after tonsillectomy.24 IgAN patients who demonstrated a significant decrease in serum GdIgA1 levels after tonsillectomy achieved significantly better improvement in hematuria, strongly indicating that at least some factors associated with nephritogenic GdIgA1 are delivered from the palatine tonsils.24
To determine whether GdIgA1 is the sole effector molecule of IgAN, Gharavi et al noted increased serum helix aspersa agglutinin-reactive GdIgA1 levels in patients with IgAN compared with those in their relatives; however, serum GdIgA1 levels in the relatives were still elevated compared with those in normal individuals who were not blood relatives, regardless of the absence of nephropathy.25 Thus, IgAN pathogenesis cannot be explained on the basis of elevated GdIgA1 levels alone, because other pathological factors may be involved in the continued progression of IgAN.
In addition, the concentration of immune complexes containing IgA, specifically GdIgA1, is increased in the blood and urine of IgAN patients.8,9,12,26 Serum levels of GdIgA1-IgA and GdIgA1-IgG in immune complexes are increased in IgAN.27 Suzuki et al have recently reported that the IgG autoantibodies that recognize glycan-containing epitopes on GdIgA1 exhibit unique features in the complementarity-determining region 3 of the variable region of their heavy chains.28 Furthermore, serum levels of IgG autoantibodies specific for GdIgA1 correlated with disease severity, as assessed by magnitude of proteinuria.28 The onset and progression of IgAN is believed to require GdIgA1 (first hit), as well as endogenous anti-glycan antibodies (second hit) and subsequent immune complex formation (third hit) and glomerular depositions of both (fourth hit).29
These multi-hit steps in disease progression were also reported in murine models of IgAN. IgAN-prone mice have elevated serum levels of mucosally regulated polymeric forms of IgA with aberrant oligosaccharide contents at the IgA hinge region30,31 and disease severity is not correlated with serum levels of IgA but rather with IgA immune complexes.32 Indeed, complement activity, including the lectin pathway, was strongly induced in the murine IgAN model.33
Assessment of disease diagnosis and activity using serum biomarkers based on etiology
The above-described clinical and experimental findings suggest that GdIgA1 and its related immune complexes with anti-glycan autoantibodies are essential effector molecules in IgAN pathogenesis; thus, serum levels of these molecules have shown potential as diagnostic markers for the clinical severity of IgAN.13,25,27,28
Recent studies demonstrated that increased GdIgA1 levels were associated with worsening proteinuria and a greater risk of deterioration of renal function in IgAN.34,35 In addition, the combination of high serum GdIgA1 levels and circulating levels of advanced oxidation protein products were correlated with a more rapid decline in estimated glomerular filtration rate, suggesting that oxidative stress linked to GdIgA1 may be involved in the pathogenesis of IgAN.35 Although these reports did not analyze serum IgA immune complex levels, Berthoux et al reported that serum levels of IgG and IgA autoantibodies (specific for GdIgA1) at the time of diagnostic biopsy were significantly associated with clinical progression of IgAN towards dialysis/death.36 The findings of previous reports further confirmed the multi-hit hypothesis for the disease mechanism of IgAN,29 and indicate the possibility that evaluation of not only serum levels of the autoantigen (GdIgA1) but also those of autoantibodies to GdIgA1 or immune complexes with such antibodies27,28 should be required as disease markers for IgAN.
Our recent study further demonstrated the value of these biomarkers for the assessment of disease activity.37 We evaluated changes in serum levels of GdIgA1 and immune complexes with autoantibodies and demonstrated correlations with changes in urinary abnormalities in 50 IgAN patients, who showed complete or partial clinical remission following tonsillectomy with steroid pulse therapy before and 3–5 years after treatment. Cross-sectional analysis revealed that the degree of hematuria and proteinuria was significantly associated with serum levels of GdIgA1 and IgA immune complexes. In addition, longitudinal analyses showed that improvement of urinary abnormalities was well correlated with decreased serum levels of GdIgA1 and IgA immune complex from baseline values before treatment. Because the disease course of IgAN is slowly progressive, clinical studies to investigate the efficacy of therapy with hard end-point evaluations as observed in diabetic nephropathy, will take a long time to complete. However, these findings emphasize the potential value of these effector molecules as practical surrogate markers for evaluation of the efficacy of therapies for IgAN.
Higher serum levels of GdIgA1 and related immune complexes indicate their diagnostic value for IgAN.13,25,27,28 However, previous studies have also shown elevated serum levels of GdIgA1 and immune complexes in some healthy individuals as well as other chronic kidney disease patients.13,14,25,27,28 In our analysis, 41% of IgAN patients had elevated serum GdIgA1 levels and 91% of these patients exhibited GdIgA1-specific IgG levels above the 90th percentile of healthy controls,38 consistent with a prior report.39 Although up to 25% of controls with chronic kidney disease, particularly those with immune-mediated glomerular diseases including lupus nephritis, also had elevated serum levels of GdIgA1-specific IgG, most IgAN patients had elevated levels of Gd-IgA1-specific antibody of both isotypes. Such a substantial overlap in serum levels of individual biomarkers between patients with IgAN, controls with chronic kidney disease, and healthy controls suggests that no single biomarker was sufficiently specific for IgAN.38
Serum levels of GdIgA1-specific IgG were significantly higher in IgAN patients with elevated GdIgA1 levels (P<0.001). However, approximately 91% of IgAN patients with normal serum GdIgA1 levels also exhibited elevated levels of GdIgA1-specific IgG,38 suggesting that auto-antibody production may occur independently of GdIgA1. Although serum levels of GdIgA1-specific autoantibodies have excellent predictive properties to discriminate IgAN patients from healthy controls and patients with non-immune-mediated kidney disease, the specificity is lower for discriminating IgAN patients from those with other types of immune-mediated kidney disease.38 Indeed, the assessment of GdIgA1-specific IgG showed the best performance for diagnosis of IgAN, with a sensitivity of 89% and specificity of 92%. However, these findings also suggest that a panel of such serum biomarkers may be helpful to differentiate IgAN from other glomerular diseases. In addition, other related markers, such as urinary GdIgA1 and immune complexes, should be assessed for addition to a panel in a larger cohort. Indeed, a multicenter trial of the diagnostic use of the panel with these biomarkers has already been initiated in Japan. Although their capability as prognostic markers to separate progressors and nonprogressors to end-stage kidney disease must be of keen interest, it would take more time because the prognostic value should be evaluated by more follow-up data about IgAN activity assessment with these biomarkers.
Conclusion
Specific curative treatment is required for IgAN, which is the most common form of glomerulonephritis and is associated with poor prognosis. Future research is needed to establish specific treatment regimens. However, such specific treatments may require early diagnosis of IgAN through specific targets before progression of commonly induced pathways that result in further kidney damage, such as sclerotic lesions, and an appropriate clinical trial with practical and disease-specific surrogate markers. As explained in this report, recent clinical and experimental studies emphasize that one of prospective diagnostic and disease assessment markers for IgAN are GdIgA1 and anti-glycan autoantibodies derived from the mucosa-bone marrow axis,40 and subsequent immune complexes (Figure 1). Therefore, noninvasive and real-time testing with such reasonable biomarkers on the basis of pathogenesis is critical to accomplish this goal.
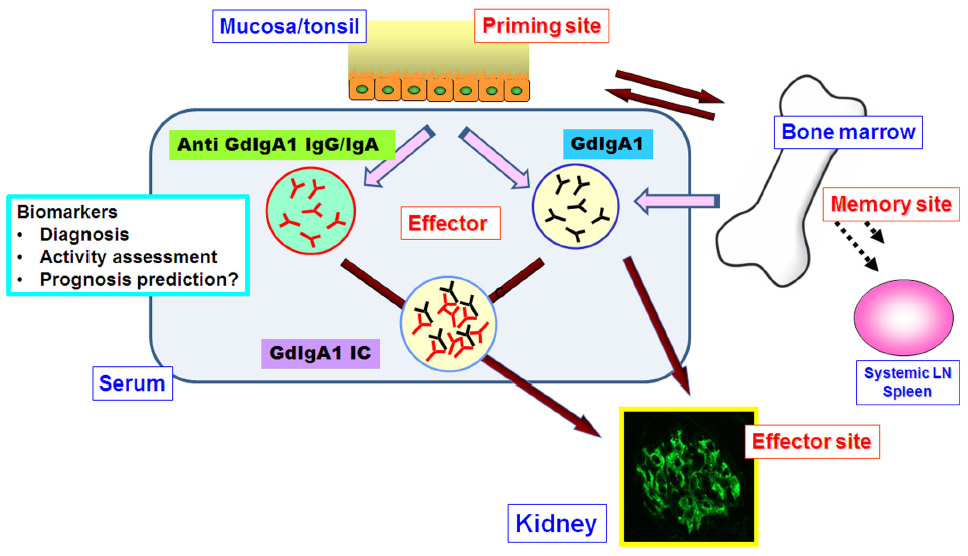 | Figure 1 GdIgA1 and its related immune complex with anti-glycan autoantibodies are prospective disease-specific surrogate markers in IgA nephropathy. Recent clinical and experimental studies show that GdIgA1 and auto anti-glycan antibodies may be derived from the mucosa bone marrow axis. |
Acknowledgments
This study was supported in part by a research grant from the Study Group on IgA Nephropathy, Grant-in-Aids for Progressive Renal Disease Research, Research on Intractable Disease and from the Ministry of Health, Labour and Welfare of Japan, and by a grant from the Strategic Japanese-Swiss Cooperative Scientific Program.
Disclosure
The authors report no conflicts of interest in this work.
References
Chauveau D, Droz D. Follow-up evaluation of the first patients with IgA nephropathy described at Necker Hospital. Contrib Nephrol. 1993;104:1–5. | |
D’Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol. 2004;24:179–196. | |
Imai H, Miura N. A treatment dilemma in adult immunoglobulin A nephropathy: what is the appropriate target, preservation of kidney function or induction of clinical remission? Clin Exp Nephrol. 2012;16:195–201. | |
Nair R, Walker PD. Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? Kidney Int. 2006;69:1455–1458. | |
Szeto CC, Lai FM, To KF, et al. The natural history of immunoglobulin: a nephropathy among patients with hematuria and minimal proteinuria. Am J Med. 2001;110:434–437. | |
Shen P, He L, Li Y, Wang Y, Chan M. Natural history and prognostic factors of IgA nephropathy presented with isolated microscopic hematuria in Chinese patients. Nephron Clin Pract. 2007;106:c157–c161. | |
Tomino Y, Endoh M, Nomoto Y, Sakai H. Immunoglobulin A1 and IgA nephropathy. N Engl J Med. 1981;305:1159–1160. | |
Barratt J, Feehally J, Smith AC. Pathogenesis of IgA nephropathy. Semin Nephrol. 2004;24:197–217. | |
Novak J, Julian BA, Mestecky J, Renfrow MB. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin Immunopathol. 2012;34:365–382. | |
Hiki Y, Odani H, Takahashi M, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. | |
Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int. 2001;60:969–973. | |
Mestecky J, Raska M, Julian BA, et al. IgA nephropathy: molecular mechanisms of the disease. Annu Rev Pathol. 2013;8:217–240. | |
Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148–1154. | |
Suzuki H, Moldoveanu Z, Hall S, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118:629–639. | |
Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest. 2014;124:2325–2332. | |
Kiryluk K, Li Y, Sanna-Cherchi S, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8:e1002765. | |
Suzuki H, Raska M, Yamada K, et al. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J Biol Chem. 2014;289:5330–5339. | |
Horie A, Hiki Y, Odani H, Yasuda Y, et al. IgA1 molecules produced by tonsillar lymphocytes are under-O-glycosylated in IgA nephropathy. Am J Kidney Dis. 2003;42:486–496. | |
Inoue T, Sugiyama H, Hiki Y, et al. Differential expression of glycogenes in tonsillar B lymphocytes in association with proteinuria and renal dysfunction in IgA nephropathy. Clin Immunol. 2010;136:447–455. | |
He L, Peng Y, Liu H, et al. Activation of the interleukin-4/signal transducer and activator of transcription 6 signaling pathway and homeodomain-interacting protein kinase 2 production by tonsillar mononuclear cells in IgA nephropathy. Am J Nephrol. 2013;38:321–332. | |
Chen X, Liu H, Peng Y, et al. Expression and correlation analysis of IL-4, IFN-γ and FcαRI in tonsillar mononuclear cells in patients with IgA nephropathy. Cell Immunol. 2014;289:70–75. | |
Sato D, Suzuki Y, Kano T, et al. Tonsillar TLR9 expression and efficacy of tonsillectomy with steroid pulse therapy in IgA nephropathy patients. Nephrol Dial Transplant. 2012;27:1090–1097. | |
Maeda A, Gohda T, Funabiki K, Horikoshi S, Shirato I, Tomino Y. Significance of serum IgA levels and serum IgA/C3 ratio in diagnostic analysis of patients with IgA nephropathy. J Clin Lab Anal. 2003;17:73–76. | |
Nakata J, Suzuki Y, Suzuki H, et al. Changes in nephritogenic serum galactose-deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLoS One. 2014;9:e89707. | |
Gharavi AG, Moldoveanu Z, Wyatt RJ, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19:1008–1014. | |
Matousovic K, Novak J, Yanagihara T, et al. IgA-containing immune complexes in the urine of IgA nephropathy patients. Nephrol Dial Transplant. 2006;21:2478–2424. | |
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. | |
Suzuki H, Fan R, Zhang Z, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119:1668–1677. | |
Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795–1803. | |
Okazaki K, Suzuki Y, Otsuji M, et al. Development of a model of early-onset IgA nephropathy. J Am Soc Nephrol. 2012;23:1364–1374. | |
Suzuki H, Suzuki Y, Narita I, et al. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19:2384–2395. | |
Suzuki H, Suzuki Y, Aizawa M, et al. Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int. 2007;72:319–327. | |
Hashimoto A, Suzuki Y, Suzuki H, et al. Determination of severity of murine IgA nephropathy by glomerular complement activation by aberrantly glycosylated IgA and immune complexes. Am J Pathol. 2012;181:1338–1347. | |
Zhao N, Hou P, Lv J, et al. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012;82:790–796. | |
Camilla R, Suzuki H, Daprà V, et al. Oxidative stress and galactose-deficient IgA1 as markers of progression in IgA nephropathy. Clin J Am Soc Nephrol. 2011;6:1903–1911. | |
Berthoux F, Suzuki H, Thibaudin L, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. 2012;23:1579–1587. | |
Suzuki Y, Matsuzaki K, Suzuki H, et al. Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin Exp Nephrol. January 30, 2014. [Epub ahead of print.] | |
Yanagawa H, Suzuki H, Suzuki Y, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One. 2014;9:e98081. | |
van der Boog PJ, van Kooten C, van Seggelen A, et al. An increased polymeric IgA level is not a prognostic marker for progressive IgA nephropathy. Nephrol Dial Transplant. 2004;19:2487–2493. | |
Suzuki Y, Tomino Y. The mucosa-bone-marrow axis in IgA nephropathy. Contrib Nephrol. 2007;157:70–79. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.