")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Developments in renal pharmacogenomics and applications in chronic kidney disease
Authors Padullés A, Rama I, Llaudó I, Lloberas N
Received 12 April 2014
Accepted for publication 16 June 2014
Published 28 August 2014 Volume 2014:7 Pages 251—266
DOI https://doi.org/10.2147/PGPM.S52763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ariadna Padullés,1 Inés Rama,2 Inés Llaudó,2 Núria Lloberas2
1Pharmacy Department, 2Nephrology Department, IDIBELL-Hospital Universitari Bellvitge, L'Hospitalet de Llobregat, Barcelona, Spain
Abstract: Chronic kidney disease (CKD) has shown an increasing prevalence in the last century. CKD encompasses a poor prognosis related to a remarkable number of comorbidities, and many patients suffer from this disease progression. Once the factors linked with CKD evolution are distinguished, it will be possible to provide and enhance a more intensive treatment to high-risk patients. In this review, we focus on the emerging markers that might be predictive or related to CKD progression physiopathology as well as those related to a different pattern of response to treatment, such as inhibitors of the renin–angiotensin system (including angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers; the vitamin D receptor agonist; salt sensitivity hypertension; and progressive kidney-disease markers with identified genetic polymorphisms). Candidate-gene association studies and genome-wide association studies have analyzed the genetic basis for common renal diseases, including CKD and related factors such as diabetes and hypertension. This review will, in brief, consider genotype-based pharmacotherapy, risk prediction, drug target recognition, and personalized treatments, and will mainly focus on findings in CKD patients. An improved understanding will smooth the progress of switching from classical clinical medicine to gene-based medicine.
Keywords: angiotensin-converting enzyme, diabetes, hypertension, renal treatment, gene polymorphisms, biomarkers
Natural history and epidemiology of chronic kidney disease (CKD)
CKD is defined as a reduced glomerular filtration rate (GFR), increased urinary albumin excretion, or both, and is a growing public health issue.1 CKD progression is related to the GFR slope or key markers of renal damage (proteinuria or albuminuria) in diabetic patients. The Kidney Disease Outcome Quality Initiative defined CKD as the presence of renal impairment with a glomerular filtration rate (GFR) <60 mL/min.2
The kidney is a key organ in the urinary system, acting as a filter of the blood, with homeostatic functions such as the regulation of electrolytes, control of blood pressure (BP), and maintenance of acid–base balance. It also modulates water imbalance, the reabsorption of several substances (such as glucose, water, and amino acids), and the excretion of urea and ammonium. Important hormones, calcitriol, erythropoietin, and the enzyme renin, are also synthesized by the kidney. In summary, kidney damage can contribute to disturbances in the equilibrium between exogenous and endogenous elements including drugs and metabolites.
CKD includes different types of renal disease. Glomerular disease is the main group, consisting of diabetic and hypertensive nephropathies, which are the leading causes of CKD in developed countries.3 Other groups of CKD, such as glomerulonephritis and CKD of unknown causes (CKDU), are more common in countries in Asia and sub-Saharan Africa, and account for 10% of CKD worldwide and 16% in India.4 Differences between countries are related mainly to chronic lifestyle-related diseases, decreased birth rates, and increased life expectancy in the developed regions. By contrast, infectious diseases continue to be prevalent in low-income countries, secondary to poor sanitation, inadequate supply of safe water, and high concentrations of disease-transmitting vectors.
There are many published studies focused on traditional risk-initiating factors such as ethnicity, sex, age, hyperfiltration, diabetes mellitus, familiar history of CKD, metabolic syndrome, albumin excretion, cardiovascular disease, primary kidney disease, and urological disorders. Progression factors such as BP, smoking, uric acid, nephrotoxins, anemia, hypertension, dyslipidemia, obesity, cardiovascular disease, proteinuria, inflammation, and hemostasis have also been evaluated.5
Aside from the conventional factors, new markers have also been implicated in CKD pathogenesis and progression; these new markers include: adiponectin, kidney injury molecule-1, liver-type fatty acid binding protein, N-terminal pro-brain natriuretic peptide, factors involved in calcium–phosphate metabolism, A-type natriuretic peptide, adrenomedullin, neutrophil gelatinase-associated lipocalin, apolipoprotein A-IV, asymmetric dimethylarginine, and some recently identified genetic polymorphisms.5
Prevalence of CKD is estimated to be 5%–16% worldwide.6,7 Complications include increased all-cause and cardiovascular mortality, kidney-disease progression, acute kidney injury, cognitive decline, anemia, mineral and bone disorders, and fractures.8–10 The Epidemiology of Chronic Kidney Disease in Spain study estimated that approximately 10% of the Spanish adult population had some degree of CKD, and similar values were also estimated from other epidemiological studies.11,12 CKD is reasonably prevalent,13 symptoms do not appear until they are at an advanced stage, and progress occurs over several years, leading to end-stage renal disease (esrD).13 In this sense, early screening of CKD would be useful to facilitate diagnosis.
The incidence of esrD differs extensively worldwide: 400 per million population per year in Taiwan; 300 per million population per year in USA and Mexico, and 100–150 per million population per year in Europe.14 Depending on the country, there are several markers and risk factors for the progression of CKD. The increasing prevalence of CKD generates concern about the cost of treatment for esrD. Bochud et al15 describe that, despite the fact that prevalence of esrD is only about 0.2%, esrD programs now account for 6.7% of total medicare expenditure, and medicare costs associated with esrD increased by 57% between 1999 and 2004.
Considering the increasing prevalence and economic impact of CKD in previous decades, genetic studies have also been used to define the gene phenotypes involved in renal impairment, such as those related to high serum creatinine levels and GFR, hypertension, diabetic nephropathy, focal segmental glomerulosclerosis, albuminuria, and esrD.17
Management issues in the treatment of CKD – clinical utility of pharmacogenomics
Here we review the link between CKD evolution and treatment, and we also identify the emerging markers and their pharmacogenomics.
Management in the treatment of CKD
The incidence of CKD, as a serious public health disease with a high morbidity and mortality, is increasing. Proteinuria is a predictor of outcome, but genetic factors have also been related to the progression of renal disease. CKD management remains a clinical challenge.
In fact, the renin–angiotensin–aldosterone system (RAAS) is a major pathway involved in the pathogenesis and progression of diabetic nephropathy,18 and the blockade of RAAS, which improves urinary protein levels, has been proven to reduce the slope of GFR in nondiabetic experimental animals and humans compared with an intensified BP control.19,20
Classical treatments
We focused on two CKD preventive and therapeutic drugs and a complicating factor in CKD that are of potential interest in kidney pharmacogenomic applications: RAAS, Vitamin D receptor (VDR) agonists and salt-sensitivity.
RAAS inhibitors
Angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) under similar conditions of blood pressure improve the progression of esrD and reduce the proteinuria rate better than non-RAAS antihypertensive drugs.18,21,22 Captopril was the first ACE inhibitor that is effective in slowing the progression of diabetic nephropathy.21 Indeed, RAAS inhibitors are considered for use in classical treatment for primary hypertension and involve two key approaches for CKD improvement, summarized by hemodynamic and antihypertensive changes as well as anti-inflammatory and antifibrotic properties. The main mechanism for both is the decrease of angiotensin II levels. With regard to inflammation, angiotensin II induces lymphocyte proliferation by nuclear factor-β (NF-kβ) activation.23 Fibrosis is attenuated by induction of extracellular-matrix proteins via transforming growth factor-β.24 On the other hand, hemodynamic beneficial effects are based on the maintenance of glomerular capillary hypertension by RAAS inhibitors.25
VDR agonists
Several metabolic disturbances of CKD, such as acidosis, dyslipidemia, and vitamin D deficiency, could also be therapeutic targets for modifying the morbidity and mortality of CKD.26
Patients with esrD show a deficiency of 1,25(OH)2D3 vitamin D and, consequently, often undergo vitamin D therapy.27 In fact, this therapy has been beneficial in hemodialysis patients in terms of survival.28 In this sense, the analogue of vitamin D has been shown to attenuate kidney interstitial fibrosis29 and ameliorate glomerulosclerosis.30 Analogues of vitamin D have also been related to a decrease in albuminuria or proteinuria in CKD.31,32
The beneficial effects of 1,25(OH)2D3 on BP and CKD progression are mediated by the NF-kβ pathway33 and by direct inhibition of 1,25(OH)2D3 on the RAAS.34,35 Both NF-kβ and RAAS are involved in immune response and related inflammation, oxidative stress, and fibrogenesis.33,36 Also, the prevention of secondary hyperparathyroidism by vitamin D treatment can ameliorate BP control.37
Salt sensitivity
Salt sensitivity of BP is still not well defined. It is accepted that a person could be considered salt-sensitive when BP increases by 5%–10% after a large increase in dietary salt intake.38 It has been described that dietary sodium intake have an impact on the efficacy of RAAS blockers in preventing CKD and cardiovascular disease.39 On the other hand, RAAS blockers’ antiproteinuric effect is impaired in patients with high sodium intake. An observational trial described that increasing sodium intake was associated with a linear increase in the risk of progression of esrD.40
Novel therapeutic approaches
Drugs focused on targeting inflammation and damaged systems (fibrosis, endothelin, oxidation, and advanced glycation end products) could be beneficial in preventing CKD progression.41
Bardoxolone methyl and palmitoylethanolamide are new drugs for the treatment of CKD that target inflammation. Bardoxolone methyl, a first-in-class oral nuclear factor erythroid 2-related factor 2 agonist, seemed to have potential as a drug for improving renal function in advance diabetic nephropathy patients in a Phase II trial. However, in the Phase III study, the treatment had to be stopped due to emerging toxicity.42,43
On the other hand, palmitoylethanolamide belongs to a fatty acid ethanolamine family, and is a new and safe nonsteroidal, anti-inflammatory, and antifibrotic agent for CKD44,45 with activity at the peroxisome proliferator-activated receptor alpha.46–48
Clinical utility of pharmacogenomics
There are numerous reasons to address the pharmacogenomics that are related to different kidney functions and treatments. Drug-treatment benefits in the pharmacogenomics of patients with kidney disease are based on avoiding nephrotoxic drugs, personalizing antihypertensive and cardiovascular drugs, and identifying the enzymes and proteins involved in the pharmacokinetics of drugs to improve renal function and BP.15 Therefore, renal pharmacogenomics encompasses three important issues: ACE inhibitors, VDR agonists, and dietary salt intake.
Forty-four genes are included in the Pharmacogenomics Knowledge Database as very important pharmacogenes for their effects on renal function and diseases.15 The most important genes involved in CKD disease are: CYP1A2 and CYP3A5; ABCB1; and methylenetetrahydrofolate reductase.
Phase I enzymes (CYP1A2 and CYP3A5)
The large interindividual variability in drug response is heritable,49,50 and single-nucleotide polymorphisms (SNPs) in genes encoding drug-metabolizing enzymes are involved in CKD.
CKDU constitutes 10% of CKD, and no specific causative agents have been identified. However, environmental toxins and heavy metals may be involved,51,52 such as persistent organic compounds that include polychlorinated biphenyls, organochlorine pesticides, and dioxins. Organic toxins are detoxified by cytochrome P450 enzymes being the CYP1A1, the enzyme that is most involved in the metabolism of persistent organic compounds. Several authors have reported the association of CYP1A1 polymorphism with various diseases such as diabetes53 and neoplasia.54–56 The prevalence of homozygous CYP1A1*2A mutants ranges between 2% and 18%, and for the heterozygous TC ranges between 32% and 55%. Siddarth et al performed a case-control study to evaluate the association of CYP1A1 in patients with CKD, and observed that subjects carrying at least one mutant allele of CYP1A1*2A (TC, CC) and *2C (AG, GG) had a 1.4- to twofold increased risk of CKDU as compared to those with the wild-type homozygous genotype, ie, TT (*2A) and AA (*2C).57 However, other studies on Indian populations observed inter- and intra-ethnic variations of these two polymorphisms.58–60
CYP1A2
CYP1A2 enzyme is responsible for 13% of the cytochrome P450 activity and has a large number of endogenous and exogenous substrates. There is a great amount of interindividual and inter-ethnic CYP1A2 variability due to both environmental and genetic factors. However, the mechanism of association between CYP1A2 and CKD is unknown, but probable mechanisms include an action mediated by CYP1A2 substrates.
Compared to other CYP-family genes, there is little data on CYP1A2 pharmacogenomics and antihypertensive drugs. The main attention has been focused on antipsychotic drugs, theophylline, and melatonin.61 Antihypertensive-drug studies have been conducted with CYP2C9, which metabolizes different antihypertensive angiotensin II receptor antagonists, such as losartan, irbesartan, candesartan, and valsartan. CYP2C9 genotype has been shown to influence losartan metabolism, and response to irbesartan differed depending on CYP2C9 genotype.62
Nutrigenomic studies have been performed to provide a mechanistic hypotheses for the relationship between CYP1A2 and BP. An increased risk of myocardial infarction with increased coffee consumption was reported among carriers of the CYP1A2 C variant.63 Regular coffee or caffeine intake increases BP,64 but there is a tolerance to the acute cardiovascular effects. There is no clear evidence that regular caffeine intake in the long-term increases the incidence of hypertension in the CYP1A2 carriers.65,66
CYP3A5
There are a large interindividual and inter-ethnic variations in CYP3A5*1 allele frequency. The CYP3A5 gene is associated with BP control, but further studies are needed to confirm the relationship with salt sensitivity in humans.67 One hypothesized mechanism is the conversion of cortisol into 6 beta-hydroxycortisol, by CYP3A5, in the kidney. However, results are not conclusive. It would be of major interest to also clarify the putative role of CYP3A5 activity on intestinal drug disposition following various dietary salt intake levels.
CYP3A4 and CYP3A5 show similar substrate specificity for each of amlodipine, felodipine, nicardipine, nifedipine, atorvastatin, pravastatin, cerivastatin, lovastatin, celiprolol, digoxin, diltiazem, enalapril, losartan, and verapamil.68,69 However, the majority of pharmacogenetic studies are concentrated on tacrolimus and cyclosporine as CYP3A5 genotypes clearly influence the pharmacokinetics of the immunosuppressant tacrolimus.70 Only a few studies with small sample sizes have analyzed the role of CYP3A5 variants on the response to drugs used to treat cardiovascular conditions. CYP3A5 variants appear to influence the pharmacokinetics of statins,71 and CYP3A5*1 carriers may experience a diminished pharmacological effect of verapamil.72 Eap et al studied the combined action of CYP3A5 and ABCB1 variants on BP, and observed that there was an association with altered response to lisinopril.73 A study of plasma amlodipine concentrations in 40 healthy Korean men observed that carriers of the CYP3A5*3/*3 genotype had lower levels of amlodipine than CYP3A5*1 carriers, but the BP decrease was similar in both groups.74
Transporters (ABCB1)
The ABCB1 gene encodes the P-glycoprotein (also named as Pgp, MDR1, and ABCB1), which belongs to the superfamily of human ABC transporters. It is also known as the multidrug resistance gene, and several ABCB1 genetic variants have been shown to influence Pgp expression in humans, including the 3435 C>T and 2677 G>T variants. Pgp is an efflux pump that transports endogenous substrates (eg, steroids, lipids, phospholipids, and cytokines), drugs (eg, digoxin, cyclosporine, tacrolimus, diltiazem, verapamil, etc), and other exogenous substrates out of the cells.75 ABCB1 polymorphisms have been widely studied in transplant patients treated with cyclosporine, and it was observed that TT carrier patients on C3435T, G2677T, and C1236T SNPs (Pgp-low pumpers) showed lower Pgp activity than noncarriers.76
SNPs related to drug transporters have also been described in CKD patients. The C3435T SNP in the gene of ABCB1 that codify P-glycoprotein was correlated with renal function and BP in two Chinese populations.77 Patients with TT genotype showed an increased risk of CKD, and higher systolic BP and pulse pressure. Results were similar in elderly subjects, with CKD with a higher risk of CKD progression and hypertension. These authors concluded the importance of ABCB1 SNP in CKD specially in elderly population. The regulation of Pgp expression seems to be influenced by multiple nuclear receptors: namely, constitutive androstane receptor-beta78 and VDR.79–81
Although the role of ABCB1 genes are widely known in the field of transplant patients, the application in CKD progression and BP regulation is still not well defined.
Nuclear receptors (VDR and PXR)
VDR is widely expressed in the human kidney, namely in the epithelial cells of the proximal and distal tubules, collecting duct, and glomerulus.82 VDR is a ligand-induced nuclear receptor that regulates the expression of over 900 genes throughout the genome,83,84 such as ABCB1,79–81 CYP24A1,84 CYP3A4,85 CYP3A7, FGF23,86 and SLC34A3. Most studies have attempted to correlate VDR polymorphisms with the development of secondary hyperparathyroidism.87 Grzegorzewska and Ostromecki described the distribution of variants of vitamin D-binding protein gene, VDR with respect to PTH serum concentrations, and response to cinacalcet treatment in patients with secondary hyperparathyroidism.88 Other studies have investigated the association of polymorphisms in the VDR gene with protection against esrD and periodontitis.89
Variants of VDR and variants within the VDR gene may influence renal function and BP, but there is a lack of conclusive data on the association with renal function. In a study of people of Indian and African descent, vitamin D deficiency was significantly associated with increased diastolic BP and triglyceride levels, and reduced high-density lipoprotein cholesterol.90 There is evidence of associations between VDR variants and diabetes, which is a major CKD risk factor. Randomized controlled trials have provided convincing evidence that VDR agonists confer renoprotection in humans.91–94
Although PXR is not currently considered to be a gene associated with BP or renal function, its role in controlling the expression of genes such as ABCB1 and CYP3A5, its involvement in steroid hormone metabolism, its action on lipid and energy metabolism, its action on inflammation, as well as its interaction with VDR all point toward PXR being an important player in kidney diseases. The role of PXR as a xenobiotic and endobiotic sensor, its ability to bind to a large array of ligands, and its numerous transcriptional gene targets suggest that PXR may mediate complex gene–environment, drug–environment and drug–drug interactions with important consequences on human health, including kidney function.
ACE gene
The ACE gene encodes ACE, an enzyme involved in the RAAS and which plays a key role in BP control. There is a high interindividual variability in circulating ACE levels, with the 287-bp Alu-repeat sequence insertion/deletion polymorphism located in intron 16 (ACE I/D) being the most extensively studied ACE genetic variant, with more than 4,000 publications during the past 20 years.95 Other ACE variants have been described, and the genetic diversity of ACE is particularly high in people of African descent.96–98 The associations of the ACE I/D polymorphism with hypertension and cardiovascular disease have been inconsistent. Some authors suggest that testing for the ACE I/D polymorphism is useful for predicting the renoprotective effect of ACE inhibitor or angiotensin-receptor blocker treatment in patients with kidney disease.99 There is currently no evidence to support a role of the ACE I/D polymorphism in predicting future risk of cardiovascular events or BP response to ACE inhibitors in the absence of renal dysfunction.
One of the first pharmacogenetic studies evaluating efficacy variability of ACE inhibitors on albumin excretion rates in nonhypertensive insulin-dependent patients with normoalbuminuria or microalbuminuria was conducted in 1998.101 The application of ACE polymorphisms has been confirmed by different authors.100 Patients carrying the II genotype had a higher albuminuria reduction and better BP control.101,102 In type I diabetic patients, the I allele had the best outcomes in terms of renal phenotypes (with decline in albuminuria and decreased BP),103–105 and type 2 diabetic patients with the II genotype plus ID alleles had decreased mortality, esrD, and diabetes progression.106 However, conflicting data exist as some other authors did not find a correlation between BP and ACE variants.107–109 ARB-treatment outcomes have also been evaluated in different studies, and the data shows that DD genotype carriers have diminished renoprotection.105,110 A recent metaregression analysis evaluated 129 papers to study the effect of ACE I/D polymorphisms on CKD risk and concluded that the D allele had the highest risk for CKD in hypertensive Asian males (odds ratio, 3.75).
Review of pathogenesis: specific genetic polymorphisms in CKD
Pathogenesis
CKD is a complex pathophysiologic process resulting from multiple etiologies. It is classified as a multifactorial disease secondary to a combination of genetic and environmental factors that influence the onset and development of esrD.111 Risk factors for development of CKD could be gathered in two groups: susceptibility to kidney disease due to sociodemographic and genetic factors; and exposure to variables that can initiate kidney disease.
The main risk factors for CKD described in the literature include hypertension, obesity, and diabetes. The global prevalence of hypertension in adults was estimated to be about 26% (972 million cases) in 2000.112 Prevalence of hypertension is higher in urban populations than in rural populations in developing countries (global prevalence is 639 million [66%]).113 The worldwide prevalence, adjusted for age and sex, is projected to increase to 1.56 billion by 2025. Therefore, treatment of hypertension is one of the most important interventions in the pharmacological management of CKD. Similar trends are apparent for diabetes and obesity.114,115 The worldwide prevalence of diabetes in adults is estimated to be 6.4%, affecting 285 million people, and is expected to rise to 7.7% by 2030 (439 million cases). The increase in overweight and obese children is particularly alarming.114 Obesity raises BP physical compression of the kidneys by increasing renal tubular sodium reabsorption, impairing pressure natriuresis, and by activating the sympathetic nervous system and RAAS.116
Genetic susceptibility is an important determining factor for the onset and/or progression of esrD and its complications, and different studies have identified new susceptibility loci for reduced renal function.117 Although environmental risk factors and interactions between genes and environment undoubtedly play an additional role, nontraditional risk factors such as oxidative stress, inflammation, and immune processes may be important contributors to the pathogenesis and progression to esrD.
Polymorphisms in CKD pathogenesis
Here we present genetic polymorphisms related to CKD and the progression to esrD (Table 1).
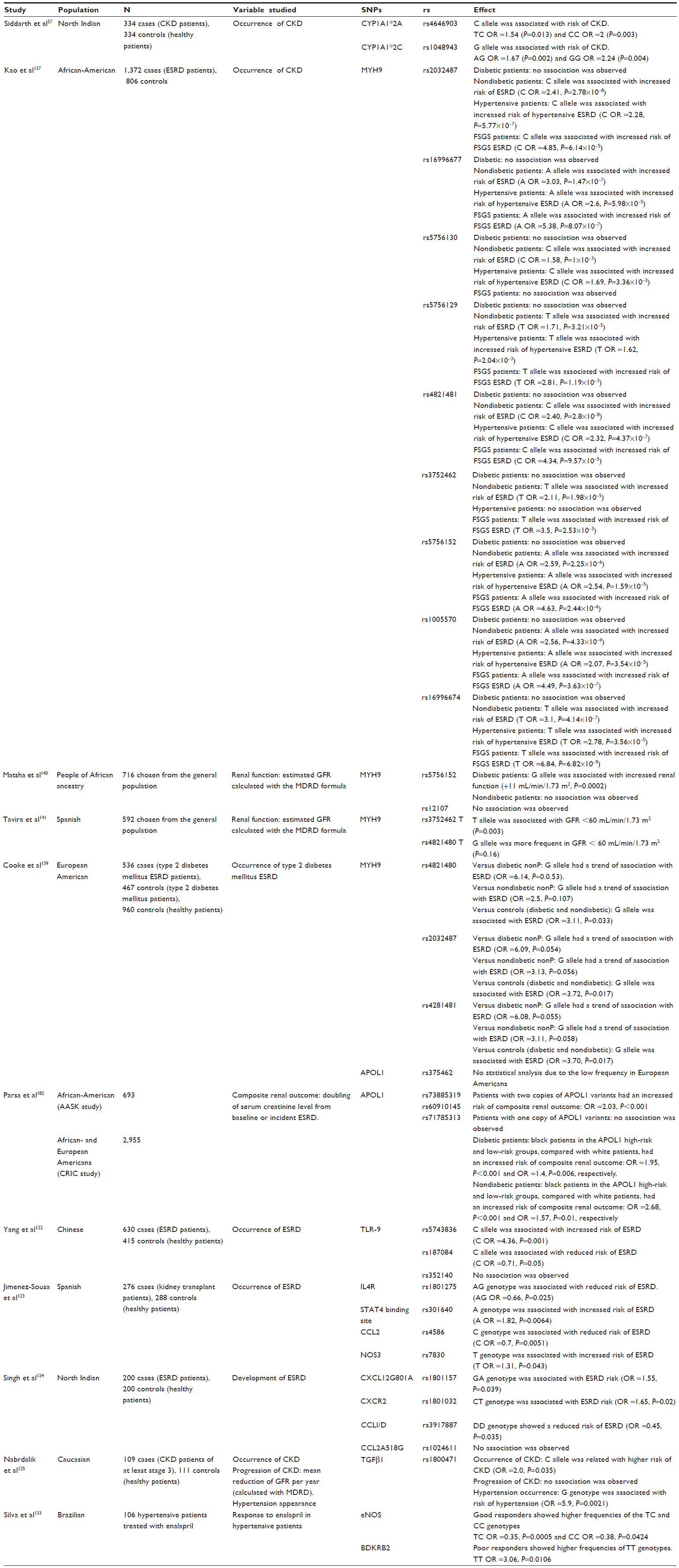 | Table 1 Characteristics and results of the main published studies involving gene polymorphisms and effect on renal function |
Inflammation could be a causal factor in the development of CKD and may be established before the onset of renal disease. The inflammatory response involved in renal damage produces proinflammatory cytokines and chemokines, an increase of leukocytes, intensification of interstitial nephritis, and a progression of fibrosis. Recent studies have suggested roles for toll-like receptor 9 (TLR-9) in the development of renal diseases such as glomerulonephritis,118 lupus nephritis,119 and the progression of immunoglobulin A nephropathy, and have also suggested that TLR-9 could be associated with severe clinical phenotypes.120,121 A case-control study observed significantly different allelic distribution of 1237T/C, but not 1486T/C or 1635G/A, between esrD patients and controls. Higher GFR values for patients with the TLR-9-1237TT genotype were obtained, but differences were not statistically significant.122
Associations of 48 chemokine gene variants with esrD have been tested; however, the small sample size did not allow to detect moderate effects. The authors found association between esrD and four SNPs.123 Two of them expressed protection (interleukin 4 receptor A/G and CCL2) and the other two expressed susceptibility (STAT4 binding site and nitric oxide synthase 3). However, after adjusting for multiple testing, the results were not significant. Singh et al found a significant association between a high risk for esrD and both CXCL2G801A and CXCR2, whereas CCL2I/D showed a reduced risk for esrD.124 In the process of CKD progression to the terminal stage, the cytokine-mediating angiotensin action (transforming growth factor-β1) could be also related as it is involved in the process of tissue sclerosis. With regard to that, Nabrdalik et al performed a case-control study and identified the mutant C allele as the related allele with the higher risk of CKD (twofold elevated risk).125 These results were similar to those observed by Buraczynska et al and Coll et al.126,127 In contrast, other authors did not find an association between transforming growth factor-β1 and CKD occurrence.128–130
Hypertension is second to diabetes as the leading independent cause of esrD. There is available evidence supporting the association of genetic variants of the RAAS and pharmacogenetic responses. ACE gene polymorphism (ACE-ID) has been associated with higher circulating plasma ACE concentrations,131 and the molecular variant M235T of the AGT gene has been associated with higher plasma AGT levels in patients homozygous for the T allele.132 Polymorphisms related to treatment efficacy with ACE inhibitors and angiotensin II receptor blockers (ARBs) are explained in Table 1. Several studies have linked variants of AGTR1 with hypertension and heart diseases, but conclusive data related to esrD is lacking. The reduction of BP by ACE inhibitors is mediated by decreased formation of the vasoconstrictor angiotensin II, and by increased levels of the vasodilator bradykinin and endothelial nitric oxide synthase (eNOS). Bradykinin receptor B2 and eNos gene polymorphism could affect the response to ACE inhibitors, and was studied by Silva et al.133 The results showed that the C allele for eNOS and TT genotype for the bradykinin receptor B2 were more frequent in good responders.
The MYH9 gene encodes for the non-muscle myosin IIA heavy chain, a subunit of myosin IIA protein that is involved in several functions including cytokinesis, cell motility, maintenance of cell shape, and secretion. Recent studies have linked MYH9 SNPs/haplotypes to a risk of developing focal segmental glomerulosclerosis, hypertensive nephropathy, and nondiabetic esrD among African and Hispanic Americans.134–138 Because African Americans seem to have greater risk than European Americans, even after considering socioeconomic status, a genome-wide admixture scan was performed to find genetic risk alleles. MYH9 genes were associated with two- to four-times greater risk of nondiabetic esrD. Multiple clusters of SNPs was performed and remained significant.137 As African Americans showed a higher risk for specific forms of CKD, the influence of MYH9 SNPs was studied in African ancestry populations. In diabetic participants, the G allele of rs5756152 was associated with a significant decrease in serum creatinine and GFR, but not in nondiabetic patients. Interactions by diabetic status were also significant, but rs12107 analysis showed no association with renal function.140
In a Spanish cohort of healthy elderly individuals, Tavira et al studied the effect of common MYH9 SNPs on renal function. The multivariate analysis showed that age, diabetes, sex, and the MYH9 genotype were risk factors for GFR <60 mL/min/1.73 m2. MYH9 rs3752462 T carriers had lower mean GFR compared to CC homozygotes.141 Aside from MYH9 polymorphisms, variations in the gene encoding apolipoprotein L1 (APOL1) have recently been shown to be associated with kidney disease in African Americans.142–145 Fifteen MYH9 SPNs and two APOL1 SNPs were studied in association with type 2 diabetes mellitus in European Americans. As reported previously, MYH9 polymorphisms were associated with type 2 diabetes mellitus esrD susceptibility, and APOL1 could not be tested for as the frequency was not appreciable.139 APOL1 variants have previously been studied and it was concluded that these polymorphisms were not present in European Americans or in Europeans.146 Polymorphisms in APOL1 related with CKD progression has been evaluated in two populations: black African Americans with kidney disease and hypertension (African American Study of Kidney Disease and Hypertension); and black and white African and European Americans with chronic renal insufficiency (Chronic Renal Insufficiency Cohort study). Black subjects in the APOL1 high-risk group had a more rapid decline in GFR and a higher risk of composite renal outcome, defined as doubling serum creatinine or incident of esrD, compared to white patients.
Developments in pharmacogenomics – clinical implications and personalized therapies
In the last few decades, despite the fact that CKD diagnosis and progression risks are still based on clinical observations, there have been notable advances in genetic studies that have led to a more selective set of therapeutic strategies.147 Herein, we review some of the potential applications of genotyping that have benefits for CKD risk prediction, guided treatment, and guided individualized strategies.
Nowadays, diagnosis and determination of risk factors based on genotyping is available for many disorders such as breast cancer (by testing BRCA 1 and 2).150 However, in CKD patients, definitive DNA-based diagnostics and risk prediction have not yet been made available.17
However, recent studies in African-American patients of nondiabetic CKD have profiled possible risk factors in polymorphisms of apolipoprotein L-I genes136,144,145 and of the non-muscle myosin heavy chain type II isoform A.136,137
The benefits derived from pharmacogenetics are aimed at individualizing treatments, reducing drug side effects, and improving drug efficacy. For example, the effects of polymorphism of genes in the cytochrome P450 enzyme complex are widely known, and genetic tests for this polymorphism before receiving treatments are routinely practiced.151,152 Also, patients with stage 5 esrD undergoing hemodialysis may present allelic variants in the cytochrome P450 2D6 (CYP2D6), which would be determinant in the metabolism of codeine into morphine, thus pointing to different susceptibility to morphine.153 Other therapeutic applications focusing on treatment with atenolol and enalapril have been identified involving polymorphism of ACE; the suggestion of these applications is that homozygous patients with a deletion allele may benefit from other antihypertensive drugs.154 These novel applications in pharmacogenomics that involve personalized treatment of patients with CKD provide clinicians with key management tools that are far from classical treatments.
Studying genetic variants may be helpful for personalized therapeutic strategies. Those genetic variants that would allow prediction of risks relative to drug exposure could guide physicians in evaluating cost-effectiveness of new strategies in CKD patients. For example, deletion polymorphism in the CC-chemokine receptor 5 (CCR5) gene (CCR5Δ32), which is known to cause a dysfunctional CCR5 protein, has been related to an improvement of inflammatory state with better survival.155,156 Therefore, patients with esrD undergoing hemodialysis and, thus, suffering from a well-known persistent inflammatory state (ie, wasting syndrome) would potentially benefit from a blockade of CCR5 based therapy.18,157 Glutathione S-transferase M1 (GST M1) null allele is another polymorphism related to CKD progression caused by deletion in the GST M1 gene and related to oxidative damage protection.158,159 In fact, hemodialysis patients are exposed to a higher oxidative stress status, and it has been shown that hemodialysis patients homozygous for the GST M1 null allele had higher risk for death compared with those who possess GST M1 activity.160
Interestingly, uremic milieu (which includes oxidative stress), wasting syndrome, inflammation, anemia, vascular calcification, and concerns associated with dialysis would render patients with CKD more susceptible to genetic variants.155,161–163 Knowing these interactions of the gene environment could be helpful in clinical practice in addressing patients’ nutritional habits and lifestyle.164 In fact, renal-function impairment per se has been considered an environment factor, varying the effect of two polymorphisms of RAS (AGTR1 A1166C and ACE insertion/deletion) on left ventricular hypertrophy.165–170 Another example of gene–environment interaction that has been established is in the susceptibility of some patients to respond to ACE inhibitors depending on dietary sodium intake and the ACE deletion/deletion genotype, which would encourage the prescription of a salt-restricted diet in those patients with this genotype variant.171
It has been questioned whether epigenetics is involved in esrD progression.172 In fact, inflammation, atherosclerotic processes, and aging are related to DNA methylation and act as catalyzers in the poor prognosis of the cardiovascular disease in dialysis patients.155,161,172,173 These findings highlight the importance of targeting the epigenome with epigenetic drugs. Therapeutic applications of epigenetics in patients with CKD, and even in patients with esrD, could be focused on RNA interference, which is crucial in renal homeostasis, mainly linked to podocyte dedifferentiation-related proteinuria.174–177 Hyperhomocysteinemia has been described in CKD and esrD patients.173,178 These patients showed high levels of S-adenosylhomocysteine, causing hypomethylation of DNA.
Thus, glomerular and interstitial fibrosis could be related to epigenetic modifications trough transcriptional regulation.77,179–181 The interest in epigenetics in clarifying hypertensive and diabetic CKD is increasing.
In this sense, as CKD incidence has been increasing in recent years, utilizing the potential therapeutic benefits of interventions to prevent esrD may be helpful in reducing the economic and clinical impact of CKD.18
Conclusions
Interesting approaches to in-depth pharmacogenetics are increasing for tailored medicine. In fact, nowadays, high-risk CKD patients could be detected by genotype information. Nevertheless, these techniques assume high cost and must be optimized alongside classical clinical tests. Advances in technology in gene sequencing with epigenetic investigation, as well as well-designed studies on gene–gene interactions, gene–environment interactions, and DNA modifications (epigenetics), may improve our knowledge of CKD-related genes and subsequent patient care. These genetic markers could be useful for the prediction of CKD progression, but clinical risk factors remain more valuable in terms of prediction. However, further technical developments and epidemiological experimental data are still needed to demonstrate and establish the most cost-effective approach. The bulk of information in pharmacogenomics published nowadays is targeted at expanding this field of information, although physicians must be critical in their analysis and interpretation of the results.
Given that CKD is a complex disorder, and that it can benefit from genetic testing, further challenges include studying proteins, transcripts, and metabolites in order to correlate them with genetic data, improve clinical outcome, and lead to routine genetic tests in the clinical care of CKD.
Acknowledgments
Núria Lloberas is a researcher in ISCIII Miguel Servet (CP06/00067) and REDinREN RD12/0021/003.
Disclosure
The authors report no conflicts of interest in this work.
References
Levey AS, Atkins R, Coresh J, et al. Chronic kidney disease as a global public health problem: approaches and initiatives – a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72(3):247–259. | |
Taal MW. Chronic Kidney Disease in General Populations and Primary Care. Curr Opin Nephrol Hypertens. 2013;22(6):593–598. | |
Jha V, Garcia-Garcia G, Iseki K, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382(9888):260–272. | |
Ball S, Lloyd J, Cairns T, et al. Why is there so much end-stage renal failure of undetermined cause in UK Indo-Asians? QJM. 2001;94(4):187–193. | |
Kronenberg F. Emerging risk factors and markers of chronic kidney disease progression. Nat Rev Nephrol. 2009;5(12):677–689. | |
Hallan SI, Coresh J, Astor BC, et al. International comparison of the relationship of chronic kidney disease prevalence and ESRD risk. J Am Soc Nephrol. 2006;17(8):2275–2284. | |
Chadban SJ, Briganti EM, Kerr PG, et al. Prevalence of kidney damage in Australian adults: The AusDiab kidney study. J Am Soc Nephrol. 2003;14(7 Suppl 2):S131–S138. | |
Bowling CB, Inker LA, Gutiérrez OM, et al. Age-specific associations of reduced estimated glomerular filtration rate with concurrent chronic kidney disease complications. Clin J Am Soc Nephrol. 2011;6(12):2822–2828. | |
Dalrymple LS, Mohammed SM, Mu Y, et al. Risk of cardiovascular events after infection-related hospitalizations in older patients on dialysis. Clin J Am Soc Nephrol. 2011;6(7):1708–1713. | |
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004;351(13):1296–1305. | |
Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298(17):2038–2047. | |
Otero A, de Francisco A, Gayoso P, García F, EPIRCE Study Group. Prevalence of chronic renal disease in Spain: results of the EPIRCE study. Nefrologia. 2010;30(1):78–86. | |
Taal MW. Screening for chronic kidney disease: preventing harm or harming the healthy? PLoS Med. 2012;9(11):e1001345. | |
Hallan SI, Vikse BE. Relationship between chronic kidney disease prevalence and end-stage renal disease risk. Curr Opin Nephrol Hypertens. 2008;17(3):286–291. | |
Bochud M, Burnier M, Guessous I. Top Three Pharmacogenomics and Personalized Medicine Applications at the Nexus of Renal Pathophysiology and Cardiovascular Medicine. Curr Pharmacogenomics Person Med. 2011;9(4):299–322. | |
Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol. 2007;18(10):2644–2648. | |
Drawz PE, Sedor JR. The genetics of common kidney disease: a pathway toward clinical relevance. Nat Rev Nephrol. 2011;7(8):458–468. | |
Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6(6):319–330. | |
Rossing P, Hommel E, Smidt UM, Parving HH. Reduction in albuminuria predicts diminished progression in diabetic nephropathy. Kidney Int Suppl. 1994;45:S145–S149. | |
Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive treatment predicts long-term GFR decline in patients with non-diabetic renal disease. Kidney Int Suppl. 1994;45:S174–S178. | |
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329(20):1456–1462. | |
Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. | |
Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–783. | |
Ruster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17(11):2985–2991. | |
Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 2000;57(5):1803–1817. | |
Liu S, Vaziri ND. Role of PCSK9 and IDOL in the pathogenesis of acquired LDL receptor deficiency and hypercholesterolemia in nephrotic syndrome. Nephrol Dial Transplant. 2014;29(3):538–543. | |
Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–38. | |
Wolf M, Betancourt J, Chang Y, et al. Impact of activated vitamin D and race on survival among hemodialysis patients. J Am Soc Nephrol. 2008;19(7):1379–1388. | |
Tan X, Li Y, Liu Y. Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2006;17(12):3382–3393. | |
Makibayashi K, Tatematsu M, Hirata M, et al. A vitamin D analog ameliorates glomerular injury on rat glomerulonephritis. Am J Pathol. 2001;158(5):1733–1741. | |
Fishbane S, Chittineni H, Packman M, Dutka P, Ali N, Durie N. Oral paricalcitol in the treatment of patients with CKD and proteinuria: a randomized trial. Am J Kidney Dis. 2009;54(4):647–652. | |
Szeto CC, Chow KM, Kwan BC, Chung KY, Leung CB, Li PK. Oral calcitriol for the treatment of persistent proteinuria in immunoglobulin A nephropathy: an uncontrolled trial. Am J Kidney Dis. 2008;51(5):724–731. | |
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–288. | |
Resnick LM, Müller FB, Laragh JH. Calcium-regulating hormones in essential hypertension. Relation to plasma renin activity and sodium metabolism. Ann Intern Med. 1986;105(5):649–654. | |
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–238. | |
Guijarro C, Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 2001;59(2):415–424. | |
Richart T, Li Y, Staessen JA. Renal versus extrarenal activation of vitamin D in relation to atherosclerosis, arterial stiffening, and hypertension. Am J Hypertens. 2007;20(9):1007–1015. | |
Sanada H, Jones JE, Jose PA. Genetics of salt-sensitive hypertension. Curr Hypertens Rep. 2011;13(1):55–66. | |
Lambers Heerspink HJ, Holtkamp FA, Parving HH, et al. Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int. 2012;82(3):330–337. | |
Vegter S, Perna A, Postma MJ, Navis G, Remuzzi G, Ruggenenti P. Sodium intake, ACE inhibition, and progression to ESRD. J Am Soc Nephrol. 2012;23(1):165–173. | |
Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. Kidney Int. 2012;81(4):351–362. | |
Pergola PE, Krauth M, Huff JW, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am J Nephrol. 2011;33(5):469–476. | |
Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365(4):327–336. | |
Indraccolo U, Barbieri F. Effect of palmitoylethanolamide-polydatin combination on chronic pelvic pain associated with endometriosis: preliminary observations. Eur J Obstet Gynecol Reprod Biol. 2010;150(1):76–79. | |
Truini A, Biasiotta A, Di Stefano G, et al. Palmitoylethanolamide restores myelinated-fibre function in patients with chemotherapy-induced painful neuropathy. CNS Neurol Disord Drug Targets. 2011;10(8):916–920. | |
Yoshihara D, Kugita M, Yamaguchi T, et al. Global Gene Expression Profiling in PPAR-γ Agonist-Treated Kidneys in an Orthologous Rat Model of Human Autosomal Recessive Polycystic Kidney Disease. PPAR Res. 2012;2012:695898. | |
Park CW, Zhang Y, Zhang X, et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69(9):1511–1517. | |
Merk D, Schubert-Zsilavecz M. New hope or drawbacks: will chronic kidney disease be treatable with small molecules in the near future? Future Med Chem. 2012;4(3):269–271. | |
Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364(12):1144–1153. | |
Rahmioglu N, Heaton J, Clement G, et al. Genetic epidemiology of induced CYP3A4 activity. Pharmacogenet Genomics. 2011;21(10):642–651. | |
Soderland P, Lovekar S, Weiner DE, Brooks DR, Kaufman JS. Chronic kidney disease associated with environmental toxins and exposures. Adv Chronic Kidney Dis. 2010;17(3):254–264. | |
Rajapurkar MM, John GT, Kirpalani AL, et al. What do we know about chronic kidney disease in India: first report of the Indian CKD registry. BMC Nephrol. 2012;13:10. | |
Wang XL, Greco M, Sim AS, Duarte N, Wang J, Wilcken DE. Effect of CYP1A1 MspI polymorphism on cigarette smoking related coronary artery disease and diabetes. Atherosclerosis. 2002;162(2):391–397. | |
Sergentanis TN, Economopoulos KP. Four polymorphisms in cytochrome P450 1A1 (CYP1A1) gene and breast cancer risk: a meta-analysis. Breast Cancer Res Treat. 2010;122(2):459–469. | |
Wright CM, Larsen JE, Colosimo ML, et al. Genetic association study of CYP1A1 polymorphisms identifies risk haplotypes in nonsmall cell lung cancer. Eur Respir J. 2010;35(1):152–159. | |
Chen J, Cheng M, Yi L, Jiang CB. Relationship between CYP1A1 genetic polymorphisms and renal cancer in China. Asian Pac J Cancer Prev. 2011;12(9):2163–2166. | |
Siddarth M, Datta SK, Ahmed RS, Banerjee BD, Kalra OP, Tripathi AK. Association of CYP1A1 gene polymorphism with chronic kidney disease: a case control study. Environ Toxicol Pharmacol. 2013;36(1):164–170. | |
Sobti RC, Sharma S, Joshi A, Jindal SK, Janmeja A. Genetic polymorphism of the CYP1A1, CYP2E1, GSTM1 and GSTT1 genes and lung cancer susceptibility in a North Indian population. Mol Cell Biochem. 2004;266(1–2):1–9. | |
Sreeja L, Syamala V, Hariharan S, Madhavan J, Devan SC, Ankathil R. Possible risk modification by CYP1A1, GSTM1 and GSTT1 gene polymorphism in lung cancer susceptibility in a south Indian population. J Hum Genet. 2005;50(12):618–627. | |
Anantharaman D, Chaubal PM, Kannan S, Bhisey RA, Mahimkar MB. Susceptibility to oral cancer by genetic polymorphisms at CYP1A1, GSTM1 and GSTT1 loci among Indians: tobacco exposure a risk modulator. Cacinogenesis. 2007;28(7):1455–1462. | |
Gunes A, Dahl ML. Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms. Pharmacogenomics. 2008;9(5):625–637. | |
Hallberg P, Karlsson J, Kurland L, et al. The CYP2C9 genotype predicts the blood pressure response to irbesartan: results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation vs Atenolol (SILVHIA) trial. J Hypertens. 2002;20(10):2089–2093. | |
Cornelis MC, Monda KL, Yu K, et al. Genome-wide meta-analysis identifies regions on 7p21 (AHR) and 15q24 (CYP1A2) as determinants of habitual caffeine consumption. PLoS Genet. 2011;7(4):e1002033. | |
Noordzij M, Uiterwaal CS, Arends LR, Kok FJ, Grobbee DE, Geleijnse JM. Blood pressure response to chronic intake of coffee and caffeine: a meta-analysis of randomized controlled trials. J Hypertens. 2005;23(5):921–928. | |
Klag MJ, Wang NY, Meoni LA, et al. Coffee intake and risk of hypertension: the Johns Hopkins precursors study. Arch Intern Med. 2002;162(6):657–662. | |
Winkelmayer WC, Stampfer MJ, Willett WC, Curhan GC. Habitual caffeine intake and the risk of hypertension in women. JAMA. 2005;294(18):2330–2335. | |
Bochud M, Bovet P, Burnier M, Eap CB. CYP3A5 and ABCB1 genes and hypertension. Pharmacogenomics. 2009;10(3):477–487. | |
Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21):2211–2221. | |
Siest G, Jeannesson E, Visvikis-Siest S. Enzymes and pharmacogenetics of cardiovascular drugs. Clin Chim Acta. 2007;381(1):26–31. | |
Daly AK. Significance of the minor cytochrome P450 3A isoforms. Clin Pharmacokinet. 2006;45(1):13–31. | |
Willrich MA, Hirata MH, Hirata RD. Statin regulation of CYP3A4 and CYP3A5 expression. Pharmacogenomics. 2009;10(6):1017–1024. | |
Jin Y, Wang YH, Miao J, et al. Cytochrome P450 3A5 genotype is associated with verapamil response in healthy subjects. Clin Pharmacol Ther. 2007;82(5):579–585. | |
Eap CB, Bochud M, Elston RC, et al. CYP3A5 and ABCB1 genes influence blood pressure and response to treatment, and their effect is modified by salt. Hypertension. 2007;49(5):1007–1014. | |
Kim KA, Park PW, Lee OJ, et al. Effect of CYP3A5*3 genotype on the pharmacokinetics and pharmacodynamics of amlodipine in healthy Korean subjects. Clin Pharmacol Ther. 2006;80(6):646–656. | |
Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34(1–2):47–54. | |
Llaudó I, Colom H, Giménez-Bonafé P, et al. Do drug transporter (ABCB1) SNPs and P-glycoprotein function influence cyclosporine and macrolides exposure in renal transplant patients? Results of the pharmacogenomic substudy within the symphony study. Transpl Int. 2013;26(2):177–186. | |
Liu M, Li Y, Citterio L, et al. A functional common polymorphism of the ABCB1 gene is associated with chronic kidney disease and hypertension in Chinese. Am J Hypertens. 2013;26(12):1428–1436. | |
Burk O, Arnold KA, Geick A, Tegude H, Eichelbaum M. A role for constitutive androstane receptor in the regulation of human intestinal MDR1 expression. Biol Chem. 2005;386(6):503–513. | |
Tachibana S, Yoshinari K, Chikada T, Toriyabe T, Nagata K, Yamazoe Y. Involvement of Vitamin D receptor in the intestinal induction of human ABCB1. Drug Metab Dispos. 2009;37(8):1604–1610. | |
Saeki M, Kurose K, Tohkin M, Hasegawa R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem Pharmacol. 2008;76(4):531–542. | |
Chow EC, Durk MR, Cummins CL, Pang KS. 1Alpha,25-dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J Pharmacol Exp Ther. 2011;337(3):846–859. | |
Kumar R, Schaefer J, Grande JP, Roche PC. Immunolocalization of calcitriol receptor, 24-hydroxylase cytochrome P-450, and calbindin D28k in human kidney. Am J Physiol. 1994;266(3 Pt 2):F477–F485. | |
Schuster I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim Biophys Acta. 2011;1814(1):186–199. | |
Wang TT, Tavera-Mendoza LE, Laperriere D, et al. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol. 2005;19(11):2685–2695. | |
Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–1316. | |
Barthel TK, Mathern DR, Whitfield GK, et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J Steroid Biochem Mol Biol. 2007;103(3–5):381–388. | |
Santoro D, Caccamo D, Gagliostro G, et al. Vitamin D metabolism and activity as well as genetic variants of the vitamin D receptor (VDR) in chronic kidney disease patients. J Nephrol. 2013;26(4):636–644. | |
Grzegorzewska AE, Ostromecki G. [Gene polymorphism of the vitamin D receptor, vitamin D-binding protein and calcium-sensing receptor in respect of calcium-phosphate disturbances in chronic dialysis patients]. Przegl Lek. 2013;70(9):735–738. Polish. | |
de Souza CM, Braosi AP, Luczyszyn SM, et al. Association between vitamin D receptor gene polymorphisms and susceptibility to chronic kidney disease and periodontitis. Blood Purif. 2007;25(5–6):411–419. | |
Vélayoudom-Céphise FL, Larifla L, Donnet JP, et al. Vitamin D deficiency, vitamin D receptor gene polymorphisms and cardiovascular risk factors in Caribbean patients with type 2 diabetes. Diabetes Metab. 2011;37(6):540–545. | |
Alborzi P, Patel NA, Peterson C, et al. Paricalcitol reduces albuminuria and inflammation in chronic kidney disease: a randomized double-blind pilot trial. Hypertension. 2008;52(2):249–255. | |
Agarwal R, Acharya M, Tian J, et al. Antiproteinuric effect of oral paricalcitol in chronic kidney disease. Kidney Int. 2005;68(6):2823–2828. | |
de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010;376(9752):1543–1551. | |
Levi M. Nuclear receptors in renal disease. Biochim Biophys Acta. 2011;1812(8):1061–1067. | |
Gard PR. Implications of the angiotensin converting enzyme gene insertion/deletion polymorphism in health and disease: a snapshot review. Int J Mol Epidemiol Genet. 2010;1(2):145–157. | |
Sayed-Tabatabaei FA, Oostra BA, Isaacs A, van Duijn CM, Witteman JC. ACE polymorphisms. Circ Res. 2006;98(9):1123–1133. | |
Kramers C, Danilov SM, Deinum J, et al. Point mutation in the stalk of angiotensin-converting enzyme causes a dramatic increase in serum angiotensin-converting enzyme but no cardiovascular disease. Circulation. 2001;104(11):1236–1240. | |
Rieder MJ, Taylor SL, Clark AG, Nickerson DA. Sequence variation in the human angiotensin converting enzyme. Nat Genet. 1999;22(1):59–62. | |
Ruggenenti P, Bettinaglio P, Pinares F, Remuzzi G. Angiotensin converting enzyme insertion/deletion polymorphism and renoprotection in diabetic and nondiabetic nephropathies. Clin J Am Soc Nephrol. 2008;3(5):1511–1525. | |
Santos PC, Krieger JE, Pereira AC. Renin-angiotensin system, hypertension, and chronic kidney disease: pharmacogenetic implications. J Pharmacol Sci. 2012;120(2):77–88. | |
Penno G, Chaturvedi N, Talmud PJ, et al. Effect of angiotensin-converting enzyme (ACE) gene polymorphism on progression of renal disease and the influence of ACE inhibition in IDDM patients: findings from the EUCLID Randomized Controlled Trial. EURODIAB Controlled Trial of Lisinopril in IDDM. Diabetes. 1998;47(9):1507–1511. | |
Ueda S, Meredith PA, Morton JJ, Connell JM, Elliott HL. ACE (I/D) genotype as a predictor of the magnitude and duration of the response to an ACE inhibitor drug (enalaprilat) in humans. Circulation. 1998;98(20):2148–2153. | |
Jacobsen P, Rossing K, Rossing P, et al. Angiotensin converting enzyme gene polymorphism and ACE inhibition in diabetic nephropathy. Kidney Int. 1998;53(4):1002–1006. | |
Jacobsen P, Tarnow L, Carstensen B, Hovind P, Poirier O, Parving HH. Genetic variation in the Renin-Angiotensin system and progression of diabetic nephropathy. J Am Soc Nephrol. 2003;14(11):2843–2850. | |
Parving HH, de Zeeuw D, Cooper ME, et al. ACE gene polymorphism and losartan treatment in type 2 diabetic patients with nephropathy. J Am Soc Nephrol. 2008;19(4):771–779. | |
So WY, Ma RC, Ozaki R, et al. Angiotensin-converting enzyme (ACE) inhibition in type 2, diabetic patients – interaction with ACE insertion/deletion polymorphism. Kidney Int. 2006;69(8):1438–1443. | |
Filigheddu F, Argiolas G, Bulla E, et al. Clinical variables, not RAAS polymorphisms, predict blood pressure response to ACE inhibitors in Sardinians. Pharmacogenomics. 2008;9(10):1419–1427. | |
Yu H, Zhang Y, Liu G. Relationship between polymorphism of the angiotensin-converting enzyme gene and the response to angiotensin-converting enzyme inhibition in hypertensive patients. Hypertens Res. 2003;26(11):881–886. | |
Arnett DK, Davis BR, Ford CE, et al. Pharmacogenetic association of the angiotensin-converting enzyme insertion/deletion polymorphism on blood pressure and cardiovascular risk in relation to antihypertensive treatment: the Genetics of Hypertension-Associated Treatment (GenHAT) study. Circulation. 2005;111(25):3374–3383. | |
Nonoguchi H, Nakayama Y, Shiigai T, et al. Low-responders to angiotensin II receptor blockers and genetic polymorphism in angiotensin-converting enzyme. Clin Nephrol. 2007;68(4):209–215. | |
Adler S. Renal disease: environment, race, or genes? Ethn Dis. 2006;16(2 Suppl 2):S2-35-39. | |
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–223. | |
Ibrahim MM, Damasceno A. Hypertension in developing countries. Lancet. 2012;380(9841):611–619. | |
Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87(1):4–14. | |
Wang Y, Mi J, Shan XY, Wang QJ, Ge KY. Is China facing an obesity epidemic and the consequences? The trends in obesity and chronic disease in China. Int J Obes (Lond). 2007;31(1):177–188. | |
Hall ME, do Carmo JM, da Silva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovasc Dis. 2014;7:75–88. | |
Köttgen A, Pattaro C, Böger CA, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42(5):376–384. | |
Summers SA, Steinmetz OM, Ooi JD, et al. Toll-like receptor 9 enhances nephritogenic immunity and glomerular leukocyte recruitment, exacerbating experimental crescentic glomerulonephritis. Am J Pathol. 2010;177(5):2234–2244. | |
Summers SA, Hoi A, Steinmetz OM, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. 2010;35(4):291–298. | |
Bochud PY, Hersberger M, Taffe P, et al. Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS. 2007;21(4):441–446. | |
Krayenbuehl PA, Hersberger M, Truninger K, et al. Toll-like receptor 4 gene polymorphism modulates phenotypic expression in patients with hereditary hemochromatosis. Eur J Gastroenterol Hepatol. 2010;22(7):835–841. | |
Yang HY, Lu KC, Lee HS, et al. Role of the functional Toll-Like receptor-9 promoter polymorphism (−1237T/C) in increased risk of end-stage renal disease: a case-control study. PLoS One. 2013;8(3):e58444. | |
Jimenez-Sousa MA, López E, Fernandez-Rodriguez A, et al. Genetic polymorphisms located in genes related to immune and inflammatory processes are associated with end-stage renal disease: a preliminary study. BMC Med Genet. 2012;13:58. | |
Singh V, Jaiswal PK, Tiwari P, Kapoor R, Mittal RD. Association of chemokine gene variants with end stage renal disease in North Indian population. Transpl Immunol. 2013;28(4):189–192. | |
Nabrdalik K, Gumprecht J, Adamczyk P, Górczynska-Kosiorz S, Zywiec J, Grzeszczak W. Association of rs1800471 polymorphism of TGFB1 gene with chronic kidney disease occurrence and progression and hypertension appearance. Arch Med Sci. 2013;9(2):230–237. | |
Buraczynska M, Baranowicz-Gaszczyk I, Borowicz E, Ksiazek A. TGF-beta1 and TSC-22 gene polymorphisms and susceptibility to microvascular complications in type 2 diabetes. Nephron Physiol. 2007;106(4):p69–p75. | |
Coll E, Cormand B, Campos B, et al. Association of TGF-beta1 polymorphisms with chronic renal disease. J Nephrol. 2004;17(6):794–799. | |
Ng DP, Warram JH, Krolewski AS. TGF-beta 1 as a genetic susceptibility locus for advanced diabetic nephropathy in type 1 diabetes mellitus: an investigation of multiple known DNA sequence variants. Am J Kidney Dis. 2003;41(1):22–28. | |
McKnight AJ, Savage DA, Patterson CC, Sadlier D, Maxwell AP. Resequencing of genes for transforming growth factor beta1 (TGFB1) type 1 and 2 receptors (TGFBR1, TGFBR2), and association analysis of variants with diabetic nephropathy. BMC Med Genet. 2007;8:5. | |
Babel N, Gabdrakhmanova L, Hammer MH, et al. Predictive value of cytokine gene polymorphisms for the development of end-stage renal disease. J Nephrol. 2006;19(6):802–807. | |
Yu ZY, Chen LS, Zhang LC, Zhou TB. Meta-analysis of the relationship between ACE I/D gene polymorphism and end-stage renal disease in patients with diabetic nephropathy. Nephrology (Carlton). 2012;17(5):480–487. | |
Zhou TB, Yin SS, Qin YH. Association of angiotensinogen M235T gene polymorphism with end-stage renal disease risk: a meta-analysis. Mol Biol Rep. 2013;40(2):765–772. | |
Silva PS, Fontana V, Luizon MR, et al. eNOS and BDKRB2 genotypes affect the antihypertensive responses to enalapril. Eur J Clin Pharmacol. 2013;69(2):167–177. | |
Behar DM, Rosset S, Tzur S, et al. African ancestry allelic variation at the MYH9 gene contributes to increased susceptibility to non-diabetic end-stage kidney disease in Hispanic Americans. Hum Mol Genet. 2010;19(9):1816–1827. | |
Freedman BI, Hicks PJ, Bostrom MA, et al. Non-muscle myosin heavy chain 9 gene MYH9 associations in African Americans with clinically diagnosed type 2 diabetes mellitus-associated ESRD. Nephrol Dial Transplant. 2009;24(11):3366–3371. | |
Freedman BI, Hicks PJ, Bostrom MA, et al. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int. 2009;75(7):736–745. | |
Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40(10):1185–1192. | |
Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40(10):1175–1184. | |
Cooke JN, Bostrom MA, Hicks PJ, et al. Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrol Dial Transplant. 2012;27(4):1505–1511. | |
Matsha TE, Masconi K, Yako YY, et al. Polymorphisms in the non-muscle myosin heavy chain gene (MYH9) are associated with lower glomerular filtration rate in mixed ancestry diabetic subjects from South Africa. PLoS One. 2012;7(12):e52529. | |
Tavira B, Coto E, Gómez J, et al. Association between a MYH9 polymorphism (rs3752462) and renal function in the Spanish RENASTUR cohort. Gene. 2013;520(1):73–76. | |
Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21(9):1422–1426. | |
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. | |
Genovese G, Tonna SJ, Knob AU, et al. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 2010;78(7):698–704. | |
Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. 2010;128(3):345–350. | |
O’Seaghdha CM, Parekh RS, Hwang SJ, et al. The MYH9/APOL1 region and chronic kidney disease in European-Americans. Hum Mol Genet. 2011;20(12):2450–2456. | |
Witasp A, Nordfors L, Carrero JJ, et al. Genetic studies in chronic kidney disease: interpretation and clinical applicability. J Nephrol. 2012;25(6):851–864. | |
Prior TW. Carrier screening for spinal muscular atrophy. Genet Med. 2008;10(11):840–842. | |
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. | |
Robson M, Offit K. Clinical practice. Management of an inherited predisposition to breast cancer. N Engl J Med. 2007;357(2):154–162. | |
Frueh FW, Amur S, Mummaneni P, et al. Pharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug use. Pharmacotherapy. 2008;28(8):992–998. | |
Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4(10):825–833. | |
Molanaei H, Carrero JJ, Heimbürger O, et al. Influence of the CYP2D6 polymorphism and hemodialysis on codeine disposition in patients with end-stage renal disease. Eur J Clin Pharmacol. 2010;66(3):269–273. | |
van Essen GG, Rensma PL, de Zeeuw D, et al. Association between angiotensin-converting-enzyme gene polymorphism and failure of renoprotective therapy. Lancet. 1996;347(8994):94–95. | |
Carrero JJ, Qureshi AR, Parini P, et al. Low serum testosterone increases mortality risk among male dialysis patients. J Am Soc Nephrol. 2009;20(3):613–620. | |
Muntinghe FL, Verduijn M, Zuurman MW, et al. CCR5 deletion protects against inflammation-associated mortality in dialysis patients. J Am Soc Nephrol. 2009;20(7):1641–1649. | |
Muntinghe FL, Vegter S, Verduijn M, et al. Using a genetic, observational study as a strategy to estimate the potential cost-effectiveness of pharmacological CCR5 blockade in dialysis patients. Pharmacogenet Genomics. 2011;21(7):417–425. | |
Board P, Coggan M, Johnston P, Ross V, Suzuki T, Webb G. Genetic heterogeneity of the human glutathione transferases: a complex of gene families. Pharmacol Ther. 1990;48(3):357–369. | |
Ryberg D, Skaug V, Hewer A, et al. Genotypes of glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis. 1997;18(7):1285–1289. | |
Lin YS, Hung SC, Wei YH, Tarng DC. GST M1 polymorphism associates with DNA oxidative damage and mortality among hemodialysis patients. J Am Soc Nephrol. 2009;20(2):405–415. | |
Filiopoulos V, Vlassopoulos D. Inflammatory syndrome in chronic kidney disease: pathogenesis and influence on outcomes. Inflamm Allergy Drug Targets. 2009;8(5):369–382. | |
Nordfors L, Lindholm B, Stenvinkel P. End-stage renal disease – not an equal opportunity disease: the role of genetic polymorphisms. J Intern Med. 2005;258(1):1–12. | |
Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. J Intern Med. 2010;268(5):456–467. | |
Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet. 2005;6(4):287–298. | |
Smilde TD, Asselbergs FW, Hillege HL, et al. Mild renal dysfunction is associated with electrocardiographic left ventricular hypertrophy. Am J Hypertens. 2005;18(3):342–347. | |
Smilde TD, Zuurman MW, Hillege HL, et al. Renal function dependent association of AGTR1 polymorphism (A1166C) and electrocardiographic left-ventricular hypertrophy. Am J Hypertens. 2007;20(10):1097–1103. | |
Kuznetsova T, Staessen JA, Thijs L, et al. Left ventricular mass in relation to genetic variation in angiotensin II receptors, renin system genes, and sodium excretion. Circulation. 2004;110(17):2644–2650. | |
Ohishi M, Rakugi H, Ogihara T. Association between a deletion polymorphism of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. N Engl J Med. 1994;331(16):1097–1098. | |
Schunkert H. Controversial association of left ventricular hypertrophy and the ACE I/D polymorphism – is the mist clearing up? Nephrol Dial Transplant. 1998;13(5):1109–1112. | |
Brewster UC, Perazella MA. The renin-angiotensin-aldosterone system and the kidney: effects on kidney disease. Am J Med. 2004;116(4):263–272. | |
van der Kleij FG, Schmidt A, Navis GJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism and short-term renal response to ACE inhibition: role of sodium status. Kidney Int Suppl. 1997;63:S23–S26. | |
Dwivedi RS, Herman JG, McCaffrey TA, Raj DS. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int. 2011;79(1):23–32. | |
Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation – a novel risk factor for cardiovascular disease? J Intern Med. 2007;261(5):488–499. | |
Harvey SJ, Jarad G, Cunningham J, et al. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol. 2008;19(11):2150–2158. | |
Ho JJ, Marsden PA. Dicer cuts the kidney. J Am Soc Nephrol. 2008;19(11):2043–2046. | |
Shi S, Yu L, Chiu C, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19(11):2159–2169. | |
Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol. 2008;19(11):2069–2075. | |
Perna AF, Ingrosso D, Satta E, Lombardi C, Acanfora F, De Santo NG. Homocysteine metabolism in renal failure. Curr Opin Clin Nutr Metab Care. 2004;7(1):53–57. | |
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. | |
Rashid A, Issa JP. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127(5):1578–1588. | |
Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. | |
Parsa A, Kao WH, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease.N Engl J Med. 2013;369(23):2183–2196. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.