")
Back to Journals » OncoTargets and Therapy » Volume 8
Critical appraisal of the role of ruxolitinib in myeloproliferative neoplasm-associated myelofibrosis
Authors Barosi G, Rosti V, Gale RP
Received 8 January 2015
Accepted for publication 6 March 2015
Published 18 May 2015 Volume 2015:8 Pages 1091—1102
DOI https://doi.org/10.2147/OTT.S31916
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Giovanni Barosi,1 Vittorio Rosti,1 Robert Peter Gale2
1Center for the Study of Myelofibrosis, IRCCS Policlinico S Matteo Foundation, Pavia, Italy; 2Haematology Research Centre, Division of Experimental Medicine, Department of Medicine, Imperial College London, London, UK
Abstract: The recent approval of molecular-targeted therapies for myeloproliferative neoplasm-associated myelofibrosis (MPN-MF) has dramatically changed its therapeutic landscape. Ruxolitinib, a JAK1/JAK2 tyrosine kinase inhibitor, is now widely used for first- and second-line therapy in persons with MPN-MF, especially those with disease-related splenomegaly, intermediate- or high-risk disease, and constitutional symptoms. The goal of this work is to critically analyze data supporting use of ruxolitinib in the clinical settings approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). We systematically reviewed the literature and analyzed the risk of biases in the two randomized studies (COMFORT I and COMFORT II) on which FDA and EMA approval was based. Our strategy was to apply the Grading of Recommendation, Assessment, Development and Evaluation (GRADE) approach by evaluating five dimensions of evidence: (1) overall risk of bias, (2) imprecision, (3) inconsistency, (4) indirectness, and (5) publication bias. Based on these criteria, we downgraded the evidence from the COMFORT I and COMFORT II trials for performance, attrition, and publication bias. In the disease-associated splenomegaly sphere, we upgraded the quality of evidence because of large effect size but downgraded it because of comparator choice and outcome indirectness (quality of evidence, low). In the sphere of treating persons with intermediate- or high-risk disease, we downgraded the evidence because of imprecision in effect size measurement and population indirectness. In the sphere of disease-associated symptoms, we upgraded the evidence because of the large effect size, but downgraded it because of comparator indirectness (quality of evidence, moderate). In conclusion, using the GRADE technique, we identified factors affecting the quality of evidence that were otherwise unstated. Identifying and evaluating these factors should influence the confidence with which physicians use ruxolitinib in persons with MPN-MF.
Keywords: myelofibrosis, myeloproliferative neoplasm-associated myelofibrosis, GRADE, ruxolitinib, JAK inhibitor, critical appraisal
What is known about myelofibrosis?
Myelofibrosis, better termed as myeloproliferative neoplasm-associated myelofibrosis (MPN-MF),1 is a myeloproliferative neoplasm that develops de novo (primary myelofibrosis [PMF]) or from antecedent polycythemia vera (post-PV-MF) or essential thrombocythemia (post-ET-MF). Clinical features include progressive anemia and/or splenomegaly and constitutional symptoms.2 MPN-MF is a clonal disease in which most blood cells are produced by one or a few abnormal clones. These clones have acquired somatic mutations that confer a survival advantage over normal hematopoietic cells. JAK2V617F is present in almost all persons with post-PV-MF and about 50% of persons with post-ET-MF and PMF.3–6 Persons without JAK2V617F have mutations in others genes, including CALR, TET2, and MPL.7–9
The International Prognostic Scoring System (IPSS)10 or Dynamic IPSS (DIPSS) prognostic classification11 are used to predict survival of persons with MPN-MF. Variables include hemoglobin concentration, white blood cell (WBC) level, age, percent blood blasts, and constitutional symptoms. DIPSS was recently modified into DIPSS-plus by adding three independent risk factors: platelet level, red blood cell transfusions, and cytogenetics.12 These variables are used to generate a score and sort people into risk cohorts. Median survival rates of persons in the low-, intermediate-1-, intermediate-2-, and high-risk cohorts are 15.4 years, 6.5 years, 2.9 years, and 1.3 years, respectively. Some recent data suggest that mutation state, especially CALR mutation, is an independent survival predictor.13,14
The target of conventional therapies of MPN-MF is mitigation of abnormalities in three main clinical spheres: (1) anemia, (2) splenomegaly, and (3) constitutional symptoms. Corticosteroids, danazol, erythropoietin, and immune-modulating drugs are commonly used for anemia. Hydroxyurea is often used to reduce splenomegaly, and splenectomy is sometimes considered. Constitutional symptoms are typically treated with corticosteroids. Although allotransplants cure 25%–50% of recipients with MPN-MF, they are used in less than 10% of persons.15
The diverse, overlapping, and confounded clinical spheres of MPN-MF have resulted in development of complex response-criteria definitions. In 2013, the European LeukemiaNet (ELN) and International Working Group for Myelofibrosis Research and Therapy (IWG-MRT) published response criteria designed for clinical trials.16 These recommendations are based on the notion that the definition of response should capture long-term effects of new drugs. Additionally, the Myelofibrosis Symptom Assessment Form (MF-SAF) provides a way to include a patient-oriented dimension of response.17
The decision problem
Ruxolitinib is a tyrosine kinase inhibitor of JAK1/218 that prevents activation of JAK-STAT signaling-pathway. It is thought but not proved to reduce proliferation of the MPN clone and release of inflammatory molecules. Two Phase III trials reported efficacy of ruxolitinib in persons with MPN-MF and splenomegaly or who had intermediate- or high-risk disease.9,19 These data resulted in the US Food and Drug Administration (FDA) approval of ruxolitinib in 2011 for therapy of patients with high- or intermediate-risk disease.20 In 2013, the European Medicines Agency (EMA) approved ruxolitinib for disease-related splenomegaly or symptoms. Ruxolitinib is now used as initial and later therapy for intermediate- and high-risk persons, especially those with extensive splenomegaly and/or symptoms. Some studies are being done using ruxolitinib in the allotransplant setting.21–23 A study reported efficacy of ruxolitinib in reducing spleen size and portal hypertension in persons with MPN-MF and splanchnic vein thrombosis.24
Appropriate use of ruxolitinib requires analyzing comparative efficacy and safety data. Our goal was to analyze evidence of clinical benefit of ruxolitinib in persons with MPN-MF without considering cost. Cost-effectiveness was the goal of a previous appraisal by the National Institute for Health and Care Excellence (NICE) in the UK which, in 2013, invited the manufacturer of ruxolitinib (Novartis) to submit clinical and cost-effectiveness evidence for ruxolitinib within its European-licensed indication. The NICE Appraisal Committee concluded that ruxolitinib was clinically effective but could not be considered a cost-effective use of National Health Service resources for treating disease-related splenomegaly or symptoms in adults with MPN-MF.25
Methods
We performed a structured literature search for English language publications using electronic databases such as MEDLINE (2005–2014), EMBASE (2005–2014), reviews including Cochrane Database of Systematic Reviews, and the Cochrane Controlled Trials Register. References in identified reports and reviews were screened to find additional relevant publications. Publications that measured efficacy of ruxolitinib in persons with MPN-MF with or without a comparison group were included, but open clinical trials and those published only as abstracts were excluded.
We used the Grading of Recommendation, Assessment, Development and Evaluation (GRADE) methodology to rate confidence in estimates of effect for each outcome.26 This required assessing (1) overall risk of bias, (2) imprecision, (3) inconsistency, (4) indirectness, and (5) publication bias. We explicitly used factors that increased or decreased the quality of evidence that was rated as high, moderate, low, or very low.27
Search results
Eleven publications were selected for detailed review including two randomized Phase III trials that reported short-term efficacy and safety,19,28 two studies that reported long-term efficacy and safety,29,30 and five studies that analyzed efficacy and safety across subgroups.31–36 Two Phase II studies were also reviewed in detail.37,38
COMFORT I was a double-blind, randomized, placebo-controlled Phase III trial conducted in the US, Canada, and Australia enrolling 309 subjects. Entry criteria included intermediate-2- or high-risk MPN-MF subjects with progressive disease and splenomegaly (>5 cm) who required treatment but were not candidates for conventional therapies. Subjects were randomized (1:1) to receive ruxolitinib or placebo. Subjects assigned to receive placebo could receive ruxolitinib when all randomized subjects completed 24 weeks of treatment and 50% of randomized subjects completed 36 weeks of treatment from the time of randomization or if they had a 25% increase of spleen volume. The starting ruxolitinib dose was 20 mg orally twice daily if the pretreatment platelets were >200×10E+9/L or 15 mg orally twice daily if platelets were 100–200×10E+9/L. Persons with platelets <100×10E+9/L were ineligible. During the trial, the ruxolitinib dose was adjusted according to platelet level guidelines.
The primary efficacy endpoint was the proportion of subjects with a ≥35% reduction in spleen volume from baseline at week 24 measured by magnetic resonance imaging (MRI) or computed tomography (CT) scan. A key secondary endpoint was the proportion of subjects with a ≥50% improvement (reduction) from baseline in total symptom score (TSS) at week 24 as assessed with the modified MF-SAF.36
COMFORT II was an open-label, randomized Phase III trial conducted in Europe enrolling 219 subjects with intermediate-2- or high-risk MPN-MF with splenomegaly and required treatment for symptoms. Subjects were randomized (2:1) to receive ruxolitinib or best available therapy (BAT). The primary efficacy endpoint was the proportion of subjects with a ≥35% spleen volume reduction from baseline at week 48. A key secondary endpoint was the proportion of subjects with a ≥35% spleen volume reduction from baseline at week 24.
Risks of bias in COMFORT I and COMFORT II
We first analyzed the internal validity, that is, risk of bias inherent to the trial design, of COMFORT I and COMFORT II using the Cochrane Collaboration’s risk of bias assessment tool on the domains of (1) allocation concealment, (2) blinding of subjects and personnel, (3) blinding of outcome assessment, (4) incomplete outcomes data, and (5) selective reporting.39
The main strength of these studies is their international, multicenter, randomized trial designs. Subjects in COMFORT I and COMFORT II were randomly assigned to treatment using the Interactive Voice Response System. This type of randomization is generally considered advantageous over block randomization because, with a small sample size, potential confounding variables are more likely to be evenly distributed between the cohorts than can be achieved by chance.40,41
Both studies were industry funded, COMFORT I by Incyte Corporation and COMFORT II by Novartis Pharmaceuticals. Representatives of the companies were involved in study design, data collection, interpretation, and analyses. Moreover, editorial support was provided by a medical writer funded by the companies. These factors are important when considering the risk of reporting bias. However, the authors have been transparent about conflicts of interest and have largely detailed the role of sponsors in the conduct and reporting of the trials. Furthermore, an independent Data Safety and Monitoring Board oversaw the study conduct.
To minimize biases, MRIs used to evaluate changes in spleen volume were read centrally by blinded readers. Only MRI or CT scans performed by the protocol-qualified facilities and submitted to the central imaging laboratory were evaluated. Results were not provided to investigators or the sponsor until the study was unblinded. Subjects used a handheld electronic device to record symptoms. These were date and time stamped to reduce recall bias. Substantial consideration was given to statistical methods. A power calculation to determine efficacy was performed prior to the study. Efficacy analyses were conducted on an intention-to-treat basis.
There are, however, potential performance biases in COMFORT I. Blinding was likely to be imperfect. Subjects and physicians had high probability of knowing the therapy assignment because of the dramatic effects of ruxolitinib in responders. Specifically, the rapid disappearance of symptoms, gain of body weight, increase in appetite, and even spleen size reduction (detected clinically rather than by MRI or CT scan) would be obvious to participants. Moreover, reduction in platelet count and/or appearance or worsening of anemia occurred in the majority of the patients treated with ruxolitinib targeted drug assignment. This blinding failure could result in systematic difference in factors other than the intervention of interest. For example, if physicians guessed that a subject was receiving placebo, they might be more likely to discontinue therapy. This bias could explain why at the data cutoff date the proportion of subjects who discontinued the treatment in the placebo arm was higher than in the ruxolitinib arm (24% vs 13.5%; P=0.018).
In COMFORT II, therapy assignment was not blinded. Subjects, especially responders, knew they were receiving ruxolitinib and were therefore likely to be more adherent to therapy than those receiving BAT. Because physicians also knew the therapy assignment, they might have been more likely to discontinue therapy in the BAT cohort. This bias could explain why at the data cutoff date, the proportion of subjects who discontinued the treatment in the BAT arm was higher than that of those in the ruxolitinib arm (32.8% vs 17.8%; P=0.012).
It is well known how difficult it is to blind subjects and physicians in intervention trials because of the effect of therapies, especially if one therapy is active and the second is placebo, such as in COMFORT I, or with a low or no response rate (as in COMFORT II).42–44 Consequently, when there is the possibility of this type of bias, subjects should be treated according to a strictly enforced prospectively defined protocol to ensure that interactions between subjects and physicians in both arms of the study are as similar as possible.45 This precaution was not taken in COMFORT I or COMFORT II.
Subjects enrolled in COMFORT I and COMFORT II were followed, and results reported after a median of 2 years and 3 years from randomization.29,30 Long-term analysis of randomized intervention trials is known to be subject to biases associated with a post-randomization selection for discontinuation of therapy, loss to follow-up, and nonrandom exposure to other therapies.46,47 In essence, these long-term observations are more similar to data from an observational database than a randomized trial.46 Proportions of subjects who discontinued the prescribed therapy and the reasons for discontinuation were reported for both trials. Discontinuation rates were more common in the placebo or BAT cohorts than in the ruxolitinib cohorts in both studies (Table 1). The most frequent causes for discontinuation included events potentially associated with death such as disease progression and severe adverse events. This attrition bias undermines the independent censoring assumption inherent to outcomes analyses and may distort the effect size of the outcomes.
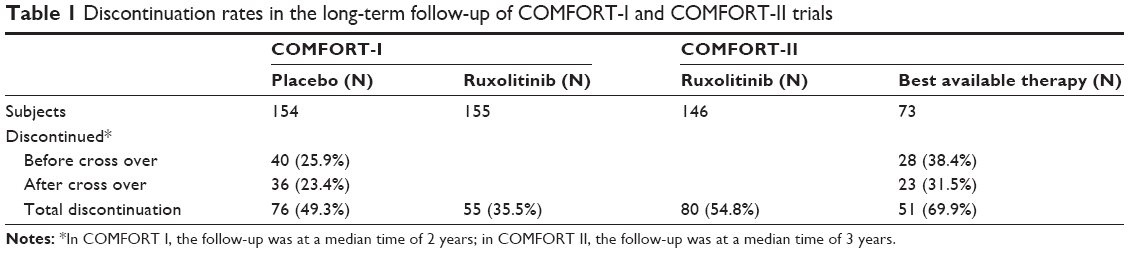 | Table 1 Discontinuation rates in the long-term follow-up of COMFORT-I and COMFORT-II trials |
Of greater importance in considering the risk of bias is the rate of loss to follow-up, since retention in care is a key measure of the success of treatment programs and greatly influences outcomes analyses. None of the publications of COMFORT I or COMFORT II cite the proportion of subjects lost to follow-up. This is an important omission.
Because of issues we discussed above, risk of bias was deemed high for both trials including performance bias, unbalanced discontinuation rates, and incomplete reporting.
Rating quality of evidence
We analyzed external validity of COMFORT I and COMFORT II by evaluating whether their results could be reasonably applied to persons with MPN-MF with specific therapeutic needs. GRADE recommends that the strength of evidence would be assessed according to a number of categorized questions (PICOs) that should include four essential constituents: (1) type of participant (P), (2) intervention (I), (3) comparator (C), and (4) outcome (O). We focused on three therapy needs: (1) relief of splenomegaly, (2) treatment of intermediate- or high-risk disease, and (3) relief of disease-associated symptoms. Selection of these therapy needs was done with consideration of the relative importance of the clinical problem, interest of hematologists, and indications approved by the US FDA and EMA. As suggested in GRADE, we evaluated four dimensions of trials quality for each question: (1) imprecision, (2) inconsistency, (3) indirectness, and (4) publication bias because these address most issues reflecting on the quality of evidence.
Role of ruxolitinib in treating splenomegaly
The population of interest, best comparator, and critical outcome relevant to persons with splenomegaly are shown in Table 2. Because not all persons with splenomegaly need therapy, we defined the population of interest by relying on expert consensus reports defining criteria for treating splenomegaly. These included (1) a spleen larger than 10 cm from the left costal margin, or a progressive splenomegaly, that is, an increase of at least 3 cm in the last year.48
 | Table 2 PICO for the role of ruxolitinib in MPN-MF-associated splenomegaly |
We analyzed the quality of evidence for COMFORT II because the study design included an active comparator (BAT). Such an analysis was not possible for COMFORT I because the comparator was placebo. At 48 weeks after randomization, 28% of subjects receiving ruxolitinib had a spleen response defined as at least 35% reduction in spleen volume from baseline. This meant that 69 out of 144 patients receiving ruxolitinib had a reduction in spleen volume of at least 35% at any time during the study, in contrast to one out of 72 of those receiving BAT. This is a response ratio of 34.5 (95% confidence interval [CI] 4.8, 243), and the lower CI boundary closest to no effect (response ratio =1) is approximately five times the control. Responses were stable; follow-up at 3 years showed that 51% of patients with post-baseline assessments achieved a spleen response according to the protocol. Thus, the evidence on the precision of the spleen response effect size after ruxolitinib was high, and we upgraded the quality of evidence.
Next, we assessed how closely subjects enrolled in COMFORT II trial resembled persons of interest, namely, what proportion of the trial subjects required therapy for splenomegaly (directness of the population) using the aforementioned consensus criteria. Eligibility criteria of COMFORT II did not include a requirement for therapy of splenomegaly. Subjects enrolled in COMFORT II had median baseline spleen measurements of 14 cm below the left costal margin (ruxolitinib cohort) and 15 cm (BAT arm). Consequently, slightly more than one-half of subjects in both cohorts met the consensus criterion for therapy for splenomegaly. However, the lower level of the measurement of spleen size at baseline was 5 cm in both arms, and there was no information about spleen progression in the preceding year. Consequently, the most conservative statistical assumption is that subjects with spleen measurement <10 cm below the left costal margin did not require therapy for splenomegaly. This inclusion of subjects not requiring therapy represents a potential indirectness of the efficacy conclusion of the relevant population.
To evaluate how this indirectness could influence confidence in the effect size measurement, we analyzed case-published series in which the enrollment criteria were closer to the consensus therapy recommendations. We analyzed effect size of a Phase II study38 enrolling 153 subjects with a spleen measurement >10 cm below the left costal margin and related symptoms. At 3 months, the response rate was 44%. These data increased the confidence that the spleen response effect size of COMFORT II is accurate and suggest that the indirectness of relevant population included in COMFORT II had a negligible impact.
A second category of potential indirectness is appropriateness of the comparator. COMFORT II compared ruxolitinib with BAT. Choosing between several BATs was left to the subject’s physician who selected a BAT after randomization using unspecified criteria. One-third of subjects in the BAT therapy received no therapy; two-thirds received anticancer drugs, most often hydroxyurea (47%). Most data suggest that the best comparators to reduce splenomegaly are hydroxyurea, busulfan, interferon, or melphalan.14 Conceivably, a substantial proportion of subjects in COMFORT II were treated for a reason(s) other than splenomegaly requiring therapy. They and others may have received a therapy, say hydroxyurea, which they previously failed. These considerations indicate indirectness of the comparator. Consequently, quality of evidence on spleen reduction effect size of ruxolitinib should be downgraded.
The primary endpoint of COMFORT II trial was a reduction of ≥35% in spleen volume from baseline at week 48 assessed by MRI. This degree of volume reduction was selected because it correlated with a 50% reduction in spleen size using clinical measurement.37 However, these endpoints are arbitrary without a clinical or biological basis. The outcome of interest in persons with splenomegaly requiring therapy is a reduction or elimination of the need for therapy. Because the IWG-MRT response criteria were formulated to address clinical relevance,16 we compared spleen response criteria of COMFORT II trial with the IWG-MRT spleen response criteria. In the IWG criteria, a clinically relevant response in a person with a spleen measurement ≥10 cm below the left costal margin is a ≥50% size reduction. A clinically relevant response in a person with a spleen measurement <10 cm below the left costal margin is a non-measurable spleen. The COMFORT II endpoint was a ≥35% spleen volume reduction even in subjects whose baseline spleen volume corresponded with a measurement <10 cm below the left costal margin. Consequently, some responders in the COMFORT II study would not be judged responders using the IWG-MRT response criteria. This discordance, reflecting indirectness of the outcome measurement in COMFORT II, could influence the confidence in the effect size measurement because a proportion of the responses were not clinically meaningful. Based on this consideration, we downgraded the quality of evidence for spleen size response in COMFORT II.
In conclusion, the two reasons for downgrading the quality of evidence on the benefit of ruxolitinib in splenomegaly requiring therapy overwhelm the one reason for upgrading the evidence for a large spleen response effect size. The sum of these considerations is to downgrade the quality of evidence for the efficacy of ruxolitinib in splenomegaly requiring therapy.
Role of ruxolitinib in intermediate- or high-risk disease
Definition of the population of interest, appropriate comparator, and critical outcome for the therapy question in persons with intermediate- or high-risk disease are shown in Table 3. Efficacy of an anticancer drug tested in a randomized trial is conventionally assessed by measuring increase in survival defined as the interval from randomization to death regardless of cause.49 COMFORT I and COMFORT II, which tested ruxolitinib against different comparators, were deemed eligible for the assessment of quality of evidence in this setting.
 | Table 3 PICO for the role of ruxolitinib in patients with intermediate- or high-risk MPN-MF |
In COMFORT I trial at a median follow-up of 32 weeks, ten deaths were reported in the ruxolitinib cohort (6.5%) as compared with 14 deaths in the placebo cohort (9.1%); hazard ratio (HR) =0.67; 95% CI 0.30–1.50; P=0.33. A subsequent planned analysis at a median follow-up of 51 weeks reported 13 deaths in the ruxolitinib cohort (8.4%) and 24 deaths in the placebo cohort (15.6%; HR =0.50; 95% CI 0.25–0.98; P=0.04). At a median 2-year follow-up, 27 deaths were reported in the ruxolitinib group and 41 in the placebo group (HR =0.58; 95% CI 0.36–0.95; P=0.03). Corresponding 24-week, 51-week, and 3-year follow-up results of the COMFORT II study are shown in Table 4. Although a survival benefit for ruxolitinib was not reported in the initial publication of the COMFORT II study, later publication based on 3-year follow-up reported better survival of subjects in the ruxolitinib cohort.30
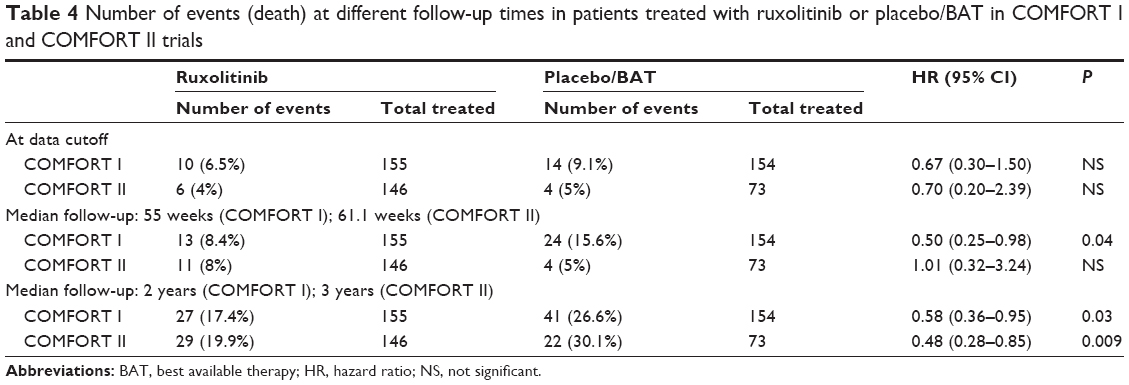 | Table 4 Number of events (death) at different follow-up times in patients treated with ruxolitinib or placebo/BAT in COMFORT I and COMFORT II trials |
We calculated pooled HRs and 95% CIs using random-effect models for all-cause mortality, given the clinical heterogeneity of COMFORT I and COMFORT II. We derived pooled estimates across the two randomized controlled trials (Figure 1), and we documented a significant benefit associated with ruxolitinib treatment at late follow-up. We used the GRADE approach to rate the confidence in estimates of effect by analyzing these results for precision. The forest plot in Figure 1 shows that the CI around the estimate of ruxolitinib’s effect on survival is in the sector of favoring better survival and providing a good measure of precision. However, this figure was obtained from trials with relatively few events, 27 in ruxolitinib cohort and 41 in the placebo cohort of COMFORT I at 2-year follow-up27 and 29 in ruxolitinib cohort and 22 in BAT cohort of COMFORT II at 3-year follow up.30 Consequently, we applied criterion of optimal information size (OIS)50 which suggests that precision is only achievable when number of subjects in a systematic review is greater than number of subjects estimated by a conventional sample size calculation for one, appropriately powered trial. We estimated considering an event rate in the control cohort of 30%, and setting an alpha of 0.05, beta of 0.02, and relative risk reduction of 25%; the result produced an OIS of 471 persons per arm or 942 total. This is almost twice as large as the 528 subjects in the combined COMFORT I and COMFORT II trials. Because our meta-analysis fails to meet OIS criteria, we downgraded confidence in the estimates of survival advantage of ruxolitinib for imprecision (too few events).
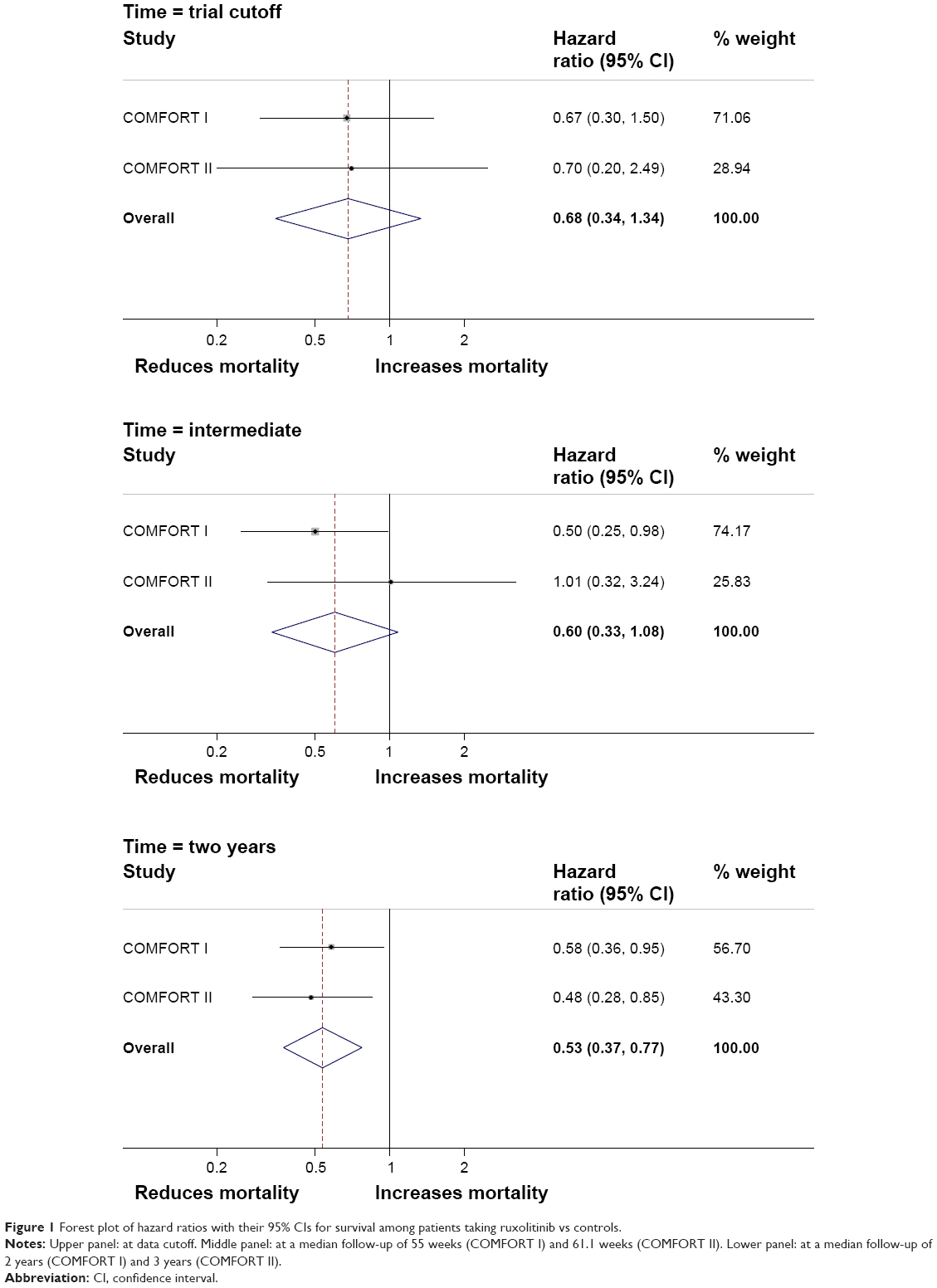 | Figure 1 Forest plot of hazard ratios with their 95% CIs for survival among patients taking ruxolitinib vs controls. |
To appraise the directness of population being considered for therapy, we assessed whether persons enrolled in COMFORT I and COMFORT II differed from persons of interest for this question. The FDA approval included patients with high- and intermediate-2-risk disease, as well as intermediate-1-risk disease, as these patients may have symptoms that require treatment. Moreover, the FDA approval of ruxolitinib did not specify an exclusion for low platelet levels even though COMFORT I and COMFORT II excluded subjects with platelets <100×10E+9/L. Thus, entry criteria of COMFORT I and COMFORT II trials differ substantially from the FDA-approved therapy indication. Because of these two issues, the risk of indirectness in the population being considered for therapy is high.
To analyze how population indirectness in COMFORT I and COMFORT II trials could influence confidence in the effect size measurement, we analyzed a Phase II trial that included subjects with platelets <100×10E+9/L.38 In this study, 50 subjects with platelets of 50–100×10E+9/L received ruxolitinib at lower doses than given in COMFORT I and COMFORT II trials. Twenty percent of subjects had a ≥35% reduction in spleen volume, a response rate lower than reported in COMFORT I and COMFORT II. This study did not report mortality and could not therefore be used to assess the potential impact of population indirectness on the precision of the survival effect size. No study has reported specifically on the survival effect size of ruxolitinib in persons with intermediate-1-risk disease.
In conclusion, the major reason for downgrading the quality of evidence supporting the use of ruxolitinib in intermediate- and high-risk disease is imprecision of the estimate of survival and the indirectness of population to be treated. Other reasons for concern over the precision of the effect size of ruxolitinib on survival are that the gain in survival in COMFORT trials became significant after 31% of subjects in COMFORT I and 52% of subjects in COMFORT II discontinued the trial for reasons other than death. As we discussed, a high level of discontinuation can result in a measured survival effect resulting from off-study therapies given after discontinuation making it difficult to assess the impact of only one therapy on survival. Lack of data on post-discontinuation therapy and data on loss to follow-up decrease confidence in survival estimates. Based on these considerations, the quality of evidence for the indication of treating intermediate- or high-risk disease was rated low.
Role of ruxolitinib in treating constitutional symptoms
Many aspects of management of MPN-MF-related constitutional symptoms remain uncertain including when to initiate therapy and what therapy(s) to use. The UK guidelines51 claim that there is no evidence of benefit for conventional drugs in this area, and they recommend that persons with severe symptoms should be considered for investigational therapies. ELN guidelines14 recommend that constitutional symptoms should be considered a key therapy indication without specifying what intervention(s) is appropriate.
A key consideration is that the lack of effective and safe therapies has influenced the therapy calculus. Ruxolitinib dramatically changes this calculus. Consequently, we framed the PICO for this question by defining the population of interest as anyone a physician considers requiring therapy for symptoms. This is obviously a subjective definition, but we know of none better (Table 5). COMFORT I and COMFORT II were judged eligible for assessing quality of evidence in this setting.
 | Table 5 PICO for the role of ruxolitinib in MPN-MF-associated symptoms |
As indicated, symptom improvement was a secondary endpoint in COMFORT I and COMFORT II. In COMFORT I, baseline TSS was calculated using an unweighted average of daily scores for a baseline week. A numerical rating scale from 0 to 10 was used, 0 indicating no symptom and 10 indicating severe symptom in six common symptom spheres: (1) night sweats, (2) itching, (3) abdominal discomfort, (4) pain under left ribs, (5) early satiety, and (6) bone/muscle pain. Mean baseline TSS was 18.0 for subjects in the ruxolitinib cohort and 16.5 for subjects in the placebo cohort. These data indicate that individual symptom scores in many subjects in COMFORT I were low. In COMFORT II, symptoms and quality of life were assessed with the use of the European Organisation for Research and Treatment of Cancer quality of life questionnaire core model and the Functional Assessment of Cancer Therapy-Lymphoma scale. A high proportion of subjects in each cohort (>95%) were evaluable for analysis of response (≥50% reduction in the TSS at week 24). Fifty percent of subjects assigned to receive ruxolitinib improved as did 5% of subjects assigned to receive placebo. The odds ratio is 15.3 (95% CI 6.9–33.7). The CI boundary closest to no effect was approximately seven times the control. In COMFORT II, the analysis was done as continuous data, and an improvement in all symptom measurements was observed. Because of the large size of the effect, we upgraded the quality of evidence on the precision of symptom benefit.
A potential bias of the studies is indirectness of the comparator. Neither placebo in COMFORT I nor BAT in COMFORT II, in which few subjects received corticosteroids, was an ideal comparator for treating disease-associated symptoms. Because of indirectness of the comparator, we judged the quality of evidence of the effect size of ruxolitinib on symptom reduction moderate.
Discussion
We present a systematic, critical appraisal of the use of ruxolitinib in MPN-associated myelofibrosis using GRADE criteria. We rated the quality of evidence supporting the proposed benefits of ruxolitinib in three key therapeutic spheres: (1) splenomegaly requiring therapy, (2) intermediate- or high-risk disease, and (3) constitutional symptoms.
We found a high risk of bias (performance bias, attrition bias, reporting bias) in the COMFORT I and COMFORT II trials that provided most of the evidence supporting efficacy of ruxolitinib in these settings. A systematic appraisal of evidence for the three therapy spheres resulted in upgrading the quality of evidence, such as large effect size of spleen volume and symptoms reduction. However, we also downgraded the quality of evidence in some areas, such as imprecision in the effect size of survival improvement, and various indirectness in the benefit in spleen volume reduction and improving symptoms. Our summary judgment is that confidence in the estimates of the effect of the ruxolitinib was low for treating persons with splenomegaly requiring therapy and for treating persons with intermediate- or high-risk disease. It was moderate for treating persons with constitutional symptoms.
The substantial risk of bias, imprecision, and indirectness in COMFORT I and COMFORT II trials seems to result from their conceptual structure. When the aim of a trial is registration of a new drug, study design and endpoints are typically negotiated with heath authorities such as the US FDA and EMA. These agencies may demand a reproducibly quantifiable measure of efficacy such as a reduction in spleen volume.52 Often the qualifier requiring therapy is included. These regulatory requirements can be problematic. For example, spleen volume measured by MRI or CT scan is not a validated endpoint of any disease-related sphere in MPN-MF. This situation is especially complex in a disease like MPN-MF in which survival is not convincingly correlated with the size of the neoplastic clone and where a substantial proportion of deaths may be unrelated to the disease or result from therapy. Many persons with MPN-MF die with rather than from the disease.
Because there is no precise, validated measure of disease severity in MPN-MF, prognostic scores such as the DIPSS correlating with survival were used to select subjects for clinical trials. These scores include some features corresponding to therapy targets (anemia, increased WBC and blasts, and constitutional symptoms) but also other variables such as age. However, it is important to consider that these scores are correlated with survival, not disease severity. As a consequence, subjects enrolled in these trials may not be representative of the universe of persons with MPN-MF who reasonably benefit from the therapy intervention. Conversely, results of the trials may not apply to many or most persons with MPN-MF.
In our opinion, using a reduction in spleen volume as primary endpoint resulted in a situation in which the trial results were not easily transferable to the specific clinical needs of most persons with MPN-MF. For example, the US FDA-approved indication of ruxolitinib is for intermediate- and high-risk disease and not splenomegaly. Thus, a drug whose therapy target is ameliorating a surrogate of symptoms (splenomegaly) is approved in an indication defined by a survival predictor (DIPSS score). This situation is referred to as a heterogeneity of ends phenomenon, a concept formulated in 1886 by the German philosopher Wilhelm Wundt. It occurs when a goal-directed activity causes experiences that modify the original motivational pattern.53 These considerations highlight debate on the adequacy of current standards for the approval of new drugs by the US FDA and EMA.54,55
There are, of course, limitations to our analysis. The dominant one is the paucity of eligible studies, resulting in a small sample size. Our appraisal of precision of effect size limiting the quality of evidence for the use of ruxolitinib in intermediate- and high-risk disease could be incorrect if the drug were given to more persons. Improving the quality of evidence requires studies with more subjects. Another limitation is that we rated quality of evidence only of efficacy outcomes. A proper application of GRADE requires a similar analysis for safety. Several adverse effects of ruxolitinib are recently reported which were not observed in COMFORT I and COMFORT II.56–65 These data suggest that the small sample size and relatively brief follow-up are not adequate for a detailed safety assessment.
The great value of our analysis is that a critical appraisal of the efficacy and safety of ruxolitinib is a prerequisite for developing clinical practice guidelines. Guidelines formulated before these analyses are complete should be viewed cautiously.
Conclusion
Splenomegaly requiring therapy and disease-associated constitutional symptoms are common in persons with MPN-MF. Ameliorating these problems can be of substantial benefit to affected persons. COMFORT I and COMFORT II and ancillary trials report sustainable efficacy and safety in these spheres. However, the large apparent reductions in spleen volume and symptoms from ruxolitinib compared with placebo or BAT likely overestimate the effect size because of the risk of biases and low-to-moderate quality of evidence. Using the GRADE approach, we uncovered factors affecting the quality of evidence of the trials that were otherwise unstated. Also, the survival benefit reported with ruxolitinib is mostly overestimated because of imprecision, indirectness, and risk of bias. A definitive analysis of whether ruxolitinib alters the natural history of MPN-MF and increases survival requires one or more large randomized trials with survival or disease-related survival as the primary or co-primary endpoints. Experts should consider issues raised in our analysis before developing guidelines for using ruxolitinib in persons with MPN-MF.
Acknowledgments
GB was supported by a grant from Associazione Italiana per la Ricerca sul Cancro (Milano) “Special Program Molecular Clinical Oncology 5x1000” to AIRC-Gruppo Italiano Malattie Mieloproliferative. RPG acknowledges support from the National Institute for Health Research Biomedical Research Centre funding scheme.
Disclosure
GB participated in advisory boards from Novartis, and was an author of COMFORT II trial publication. RPG is a part-time employee of Celgene Corp. VR reports no conflict of interest in this work.
References
Mesa RA, Green A, Barosi G, Verstovsek S, Vardiman J, Gale RP. MPN-associated myelofibrosis (MPN-MF). Leuk Res. 2011;35(1):12–13. | ||
Tefferi A. Primary myelofibrosis: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(9):915–925. | ||
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. | ||
Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. | ||
James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | ||
Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. | ||
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. | ||
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. | ||
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | ||
Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. | ||
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. | ||
Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–1810. | ||
Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494–1500. | ||
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. | ||
Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122(8):1395–1398. | ||
Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098–4103. | ||
Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Deisseroth A, Kaminskas E, Grillo J, et al. U.S. Food and Drug Administration approval: ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin Cancer Res. 2012;18(12):3212–3217. | ||
Gupta V, Gotlib J, Radich JP, et al. Janus kinase inhibitors and allogeneic stem cell transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2014;20(9):1274–1281. | ||
Jaekel N, Behre G, Behning A, et al. Allogeneic hematopoietic cell transplantation for myelofibrosis in patients pretreated with the JAK1 and JAK2 inhibitor ruxolitinib. Bone Marrow Transplant. 2014;49(2):179–184. | ||
Stübig T, Alchalby H, Ditschkowski M, et al. JAK inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post-ET/PV myelofibrosis. Leukemia. 2014;28(8):1736–1738. | ||
Pieri L, Paoli C, Arena U, et al. A phase 2 study of ruxolitinib in patients with splanchnic vein thrombosis associated with myeloproliferative neoplasm: a study from the AGIMM group. Blood. 2014; [Abstractn.72125, ASH. | ||
Wade R, Rose M, Neilson AR, et al. Ruxolitinib for the treatment of myelofibrosis: a NICE single technology appraisal. Pharmacoeconomics. 2013;31(10):841–852. | ||
Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–926. | ||
Guyatt GH, Oxman AD, Kunz R, et al. Going from evidence to recommendations. BMJ. 2008;336(7652):1049–1051. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013;98(12):1865–1871. | ||
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. | ||
Guglielmelli P, Biamonte F, Rotunno G, et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. 2014;123(14):2157–2160. | ||
Mesa RA, Shields A, Hare T, et al. Progressive burden of myelofibrosis in untreated patients: assessment of patient-reported outcomes in patients randomized to placebo in the COMFORT-I study. Leuk Res. 2013;37(8):911–916. | ||
Harrison CN, Mesa RA, Kiladjian JJ, et al. Health-related quality of life and symptoms in patients with myelofibrosis treated with ruxolitinib versus best available therapy. Br J Haematol. 2013;162(2):229–239. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br J Haematol. 2013;161(4):508–516. | ||
Mesa RA, Gotlib J, Gupta V, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2013;31(10):1285–1292. | ||
Mesa RA, Kantarjian H, Tefferi A, et al. Evaluating the serial use of the Myelofibrosis Symptom Assessment Form for measuring symptomatic improvement: performance in 87 myelofibrosis patients on a JAK1 and JAK2 inhibitor (INCB018424) clinical trial. Cancer. 2011;117(21):4869–4877. | ||
Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. | ||
Talpaz M, Paquette R, Afrin L, et al. Interim analysis of safety and efficacy of ruxolitinib in patients with myelofibrosis and low platelet counts. J Hematol Oncol. 2013;6(1):81. | ||
Higgins J, Altman D, Higgins J, Green S, editors. Chapter 8: assessing risk of bias in included studies. Cochrane Handbook for Systematic Reviews of Interventions version 5.0. Chichester, UK: John Wiley & Sons; 2008. | ||
Papaconstantinou C, Krischer JP. An automated patient registration and treatment randomization system. J Med Syst. 1995;19(6):445–456. | ||
Kuznetsova OM, Tymofyeyev Y. Expansion of the modified Zelen’s approach randomization and dynamic randomization with partial block supplies at the centers to unequal allocation. Contemp Clin Trials. 2011;32(6):962–972. | ||
Boutron I, Guittet L, Estellat C, Moher D, Hróbjartsson A, Ravaud P. Reporting methods of blinding in randomized trials assessing nonpharmacological treatments. PLoS Med. 2007;4(2):e61. | ||
Hróbjartsson A, Forfang E, Haahr MT, Als-Nielsen B, Brorson S. Blinded trials taken to the test: an analysis of randomized clinical trials that report tests for the success of blinding. Internl J Epidemiol. 2007;36(3):654–663. | ||
Wood L, Egger M, Gluud LL, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ. 2008;336(7644):601–605. | ||
Higgins JPT, Altman DG, Sterne JAC. Chapter 8: assessing risk of bias in included studies. In Higgins JPT and Green S, editors. Cochrane handbook for systematic reviews of interventions (Version 5.1.0). 2011, Retrieved from: www.cochrane-handbook.org | ||
Little RJ, D’Agostino R, Cohen ML, et al. The prevention and treatment of missing data in clinical trials. N Engl J Med. 2012;367(14):1355–1360. | ||
Hernán MA, Hernández-Díaz S, Robins JM. Randomized trials analyzed as observational studies. Ann Inten Med. 2013;159(8):500–502. | ||
Barosi G, Vannucchi AM, De Stefano V, et al. Identifying and addressing unmet clinical needs in Ph-neg classical myeloproliferative neoplasms: a consensus-based SIE, SIES, GITMO position paper. Leuk Res. 2014;38(2):155–160. | ||
Barosi G, Tefferi A, Besses C, et al. Clinical end points for drug treatment trials in BCR-ABL1-negative classic myeloproliferative neoplasms: consensus statements from European LeukemiaNET (ELN) and International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT). Leukemia. 2014;29(1):20–26. | ||
Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines 6. Rating the quality of evidence – imprecision. J Clin Epidemiol. 2011;64(12):1283–1293. | ||
Reilly JT, McMullin MF, Beer PA, et al. British Committee for Standards in Haematology. Guideline for the diagnosis and management of myelofibrosis. Br J Haematol. 2012;158(4):453–471. | ||
Liu Y, Litière S, de Vries EG, et al. The role of response evaluation criteria in solid tumour in anticancer treatment evaluation: results of a survey in the oncology community. Eur J Cancer. 2014;50(2):260–266. | ||
Wundt WM. Ethik. Eine Untersuchung der Thatsachen und Gesetze des sittlischen Lebens. Stuttgart, Germany: F. Enke; 1886. | ||
Wood AJ. A proposal for radical changes in the drug-approval process. N Engl J Med. 2006;355(6):618–623. | ||
De Palma R, Liberati A, Ciccone G, et al. Developing clinical recommendations for breast, colorectal, and lung cancer adjuvant treatments using the GRADE system: a study from the Programma Ricerca e Innovazione Emilia Romagna Oncology Research Group. J Clin Oncol. 2008;26(7):1033–1039. | ||
Lee SC, Feenstra J, Georghiou PR. Pneumocystis jiroveci pneumonitis complicating ruxolitinib therapy. BMJ Case Rep. 2014;pii: bcr2014204950. | ||
Tong LX, Jackson J, Kerstetter J, Worswick SD. Reactivation of herpes simplex virus infection in a patient undergoing ruxolitinib treatment. J Am Acad Dermatol. 2014;70(3):e59–e60. | ||
Heine A, Brossart P, Wolf D. Ruxolitinib is a potent immunosuppressive compound: is it time for anti-infective prophylaxis? Blood. 2013;122(23):3843–3844. | ||
Shen CH, Hwang CE, Chen YY, Chen CC. Hepatitis B virus reactivation associated with ruxolitinib. Ann Hematol. 2014;93(6):1075–1076. | ||
Massa M, Rosti V, Campanelli R, Fois G, Barosi G. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia. 2014;28(2):449–451. | ||
Goldberg RA, Reichel E, Oshry LJ. Bilateral toxoplasmosis retinitis associated with ruxolitinib. N Engl J Med. 2013;369(7):681–683. | ||
Caocci G, Murgia F, Podda L, Solinas A, Atzeni S, La Nasa G. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2014;28(1):225–227. | ||
Wathes R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med. 2013;369(2):197–198. | ||
Heine A, Held SA, Daecke SN, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122(7):1192–1202. | ||
Wysham NG, Sullivan DR, Allada G. An opportunistic infection associated with ruxolitinib, a novel janus kinase 1, 2 inhibitor. Chest. 2013;143(5):1478–1479. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.