")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Creutzfeldt-Jakob disease versus anti-LGI1 limbic encephalitis in a patient with progressive cognitive dysfunction, psychiatric symptoms, involuntary facio-brachio-crural movement, and an abnormal electroencephalogram: a case report
Authors Sun L, Cao J, Liu C, Lv Y
Received 23 January 2015
Accepted for publication 19 March 2015
Published 11 June 2015 Volume 2015:11 Pages 1427—1430
DOI https://doi.org/10.2147/NDT.S81414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Li Sun, Jie Cao, Chang Liu, Yudan Lv
Department of Neurology, The First Hospital of JiLin University, ChangChun, People’s Republic of China
Abstract: Diagnosis of Creutzfeldt-Jakob disease (CJD) is often challenging in elderly individuals, not only because of its variable clinical features but also because of nonspecific changes on the electroencephalogram (EEG) in the early stages of the disease. Here we report on a patient who presented with progressive cognitive dysfunction, psychiatric symptoms, involuntary facio-brachio-crural movement, and an abnormal EEG. We provide a detailed analysis and differential diagnosis between anti-leucine-rich glioma inactivated 1 (LGI1) limbic encephalitis versus CJD, in the hope of providing a new understanding of CJD. A 65-year-old Chinese man presented with slowly progressive cognitive decline with psychiatric symptoms. On admission, he presented with facial grimacing and brief left upper limb dystonic posturing lasting 1–2 seconds, with hyponatremia that was difficult to rectify. Neurological examination showed increased muscle tension in the left limb but without pathological reflexes. His early EEG showed focal periodic wave complexes. Diffusion-weighted magnetic resonance imaging showed a suspected “lace sign” in the occipital cortex. His cerebrospinal fluid was negative for LGI1 antibodies and positive for 14-3-3 brain protein. Therefore, we made a presumptive diagnosis of CJD. At the following visit, a second EEG showed paroxysmal sharp wave complexes, but the patient had a poor prognosis. Atypical facio-brachio-crural movement and nonspecific EEG changes may occasionally be found in patients with CJD or anti-LGI1 encephalitis. Clinicians should not be dissuaded from a diagnosis of CJD where the EEG does not show paroxysmal sharp wave complexes in the early stages but abnormal facio-brachio-crural movement is present.
Keywords: abnormal facio-brachio-crural movement, hyponatremia, Creutzfeldt-Jakob disease, leucine-rich glioma inactivated 1, paroxysmal sharp wave complexes
Introduction
Diagnosis of Creutzfeldt-Jakob disease (CJD)1 is often challenging in elderly individuals because the various symptoms of this condition overlap with other conditions that are common in this population, such as Alzheimer’s disease or dementia with Lewy bodies.2 However, we have had a patient who presented with atypical symptoms that were similar to those of anti-leucine-rich glioma inactivated 1 (LGI1) encephalitis. Therefore, a comprehensive understanding of CJD and anti-LGI1 encephalitis is critical, not only with regard to clinical features, but also the investigations required, which include cerebrospinal fluid (CSF), diffusion-weighted magnetic resonance imaging (MRI), and an electroencephalogram (EEG).
Here we report a case of CJD with progressive cognitive impairment, psychiatric symptoms, and complex abnormal facio-brachio-crural movement, and then present a detailed analysis, which has not been done before. Written informed consent was obtained from the patient’s son and daughter for publication of this case report and the accompanying images, and ethical approval was given by the research ethics board at the First Hospital of Jilin University.
Case report
A 65-year-old northern Chinese man developed slowly progressive cognitive decline with dizziness, and was admitted to our hospital a week after symptom onset because of unstable gait and psychiatric symptoms. At admission, neurological evaluation revealed moderate cognitive disturbance of orientation and memory, along with a Mini-Mental State Examination score of 19/30. Two days later, he developed drowsiness with facial grimacing and brief dystonic posturing of the left upper limb lasting 1–2 seconds. He was admitted to the intensive care unit, and found to have hyponatremia (serum sodium 114 mEq/L) that was difficult to rectify. There was no history of tongue bite or urinary or bowel incontinence associated with these episodes.
Neurological examination showed increased muscle tension in the left limb but no pathological reflexes. An EEG was performed to evaluate the frequently abnormal behavior of the left upper limb. His EEG video showed facial grimacing, head to the left, and external rotation of the left shoulder. The synchronized EEG was normal, with significant electromyographic artifacts, and the interictal EEG showed slow background with sharp-slow discharge in the right frontal lobe every 0.5–1.0 seconds (Figure 1). Diffusion-weighted MRI showed a suspected “lace sign” in the occipital cortex.3 His CSF study showed ten cells and normal sugar and protein levels. Tests for virus antibodies, including anti-herpes simplex, anti-rubella, anti-cytomegalovirus, and anti-Coxsackie virus, in the CSF were negative. Tests for paraneoplastic-associated antibodies in the CSF, such as anti-Ho, anti-Yo, anti-Ri, and anti-CV2, were also negative. Combining a detailed history and the EEG, MRI, and CSF findings, two diseases were considered, ie, anti-LGI1 encephalitis or CJD. The MRI and EEG results supported a diagnosis of CJD, but the hyponatremia and clinical seizures were suggestive of anti-LGI1 encephalitis. To make the differential diagnosis, we tested further for autoimmune encephalopathy, obtaining a negative result for LGI1 antibody and a positive result for 14-3-3 brain protein. During this time, the patient was not improving on immunoglobulin therapy. Therefore, we made a presumptive diagnosis of CJD according to the process together with elevated 14-3-3 protein in the CSF,4,5 but without periodic paroxysmal sharp wave complexes (PSWCs) as characteristic EEG findings.6 One week later, we performed a follow-up EEG that showed typical PSWCs (Figure 2).
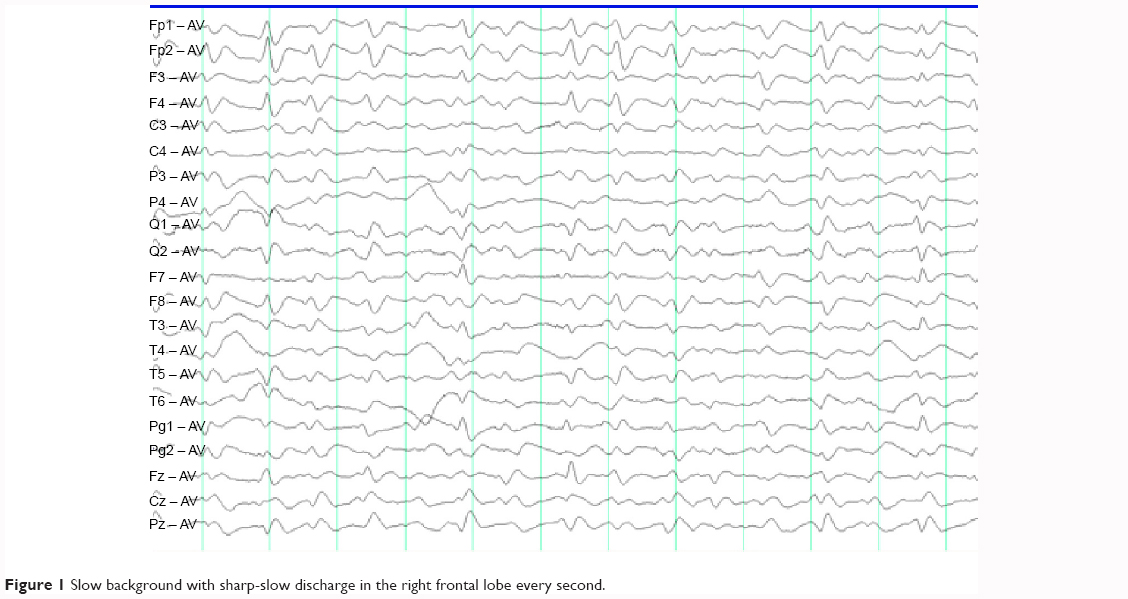 | Figure 1 Slow background with sharp-slow discharge in the right frontal lobe every second. |
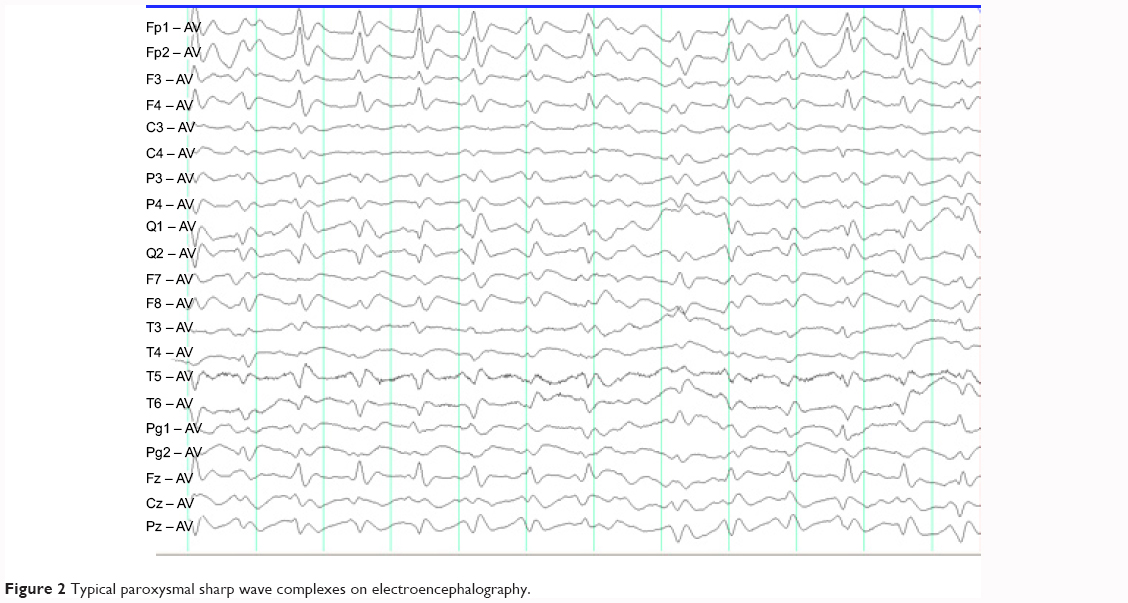 | Figure 2 Typical paroxysmal sharp wave complexes on electroencephalography. |
The patient was diagnosed as having CJD, but presented with progressive dementia, abnormal behavior including facial grimacing, external rotation of left shoulder, and hyponatremia, which is similar to the manifestations of anti-LGI1 encephalitis. In addition, in the early stages, his EEG did not show typical PSWCs, which went against a diagnosis of CJD. Therefore, atypical CJD needs a broad differential diagnosis. The patient later developed acute pancreatitis and died. The patient’s family members refused the autopsy needed for confirmation of CJD.
Discussion
Our patient’s main complaints were of cognitive impairment, psychiatric symptoms, and abnormal facio-brachio-crural movement with increased muscle tension in the left limb. However, soon after admission, he gradually lapsed into a drowsy state, with hyponatremia, a “lace sign” on MRI, and focal periodic discharges on the EEG, indicating a broad differential diagnosis, such as LGI1 or CJD. The later findings of positive 14-3-3 protein in CSF and typical PSWCs on a second EEG were helpful in the clinical diagnosis of CJD. According to the World Health Organization diagnostic criteria for probable diagnosis of CJD, the presence of at least one typical feature on the EEG and 14-3-3 positivity in CSF samples or at least two criteria of myoclonus, visual disturbances, cerebellar, pyramidal, or extrapyramidal findings, and akinetic mutism, together with progressive dementia, are required.7 Zerr et al found that 14-3-3 protein is 94% sensitive and 84% specific for CJD.8 Steinhoff et al found that periodic biphasic or triphasic synchronized sharp wave complexes had 64% sensitivity and 91% specificity for EEG examination during the intermediate or terminal phase.9 Therefore, although our patient presented with atypical abnormal facio-brachio-crural movement and hyponatremia, which was suggestive of anti-LGI1 encephalitis, he met the diagnostic criteria for CJD. Unfortunately, we were unable to perform an autopsy to confirm the diagnosis.
It is well known that PSWCs in a patient with progressive dementia suggest CJD. However, in most cases, PSWCs are absent in the initial phase, and the strict criteria for definition of PSWCs in CJD suggest a duration of 100–600 msec and an intercomplex interval of 500–2,000 msec, which may assist differentiation.6 The EEG in CJD shows different abnormalities in different phases. During the initial phase of CJD, the EEG shows a mild slowing of background activity with diffuse 5–7 Hz rhythms, and occasional bilateral or unilateral bursts of arrhythmic irregular wave complexes. During the intermediate phase, the EEG shows moderate slowing of background activity with diffuse 4–5 Hz rhythms and frequent bursts of multifocal biphasic or triphasic wave complexes. Finally, during the terminal phase, generalized PSWCs are predominant on the EEG. In the early stages, our patient’s EEG showed focal periodic wave complexes with an interval of 0.5–1.0 seconds, which was not helpful for a diagnosis of CJD. However, on progression of his illness, PSWCs were found and led to the diagnosis of CJD, so prolonged observation may be required in such cases to detect the presence of typical PSWCs on the EEG.
In addition, a new clinical syndrome of faciobrachial dystonic seizures has been characterized recently, and linked with proteins associated with the voltage-gated potassium channel.10 This syndrome is specifically associated with antibodies to LGI1 protein. This anti-LGI1 encephalitis is characterized by peculiar movement disorder and hyponatremia, which may be confused with CJD. The patient described here presented with facial grimacing and brief dystonic posturing of the left upper limb lasting 1–2 seconds, but with a normal synchronized EEG, which excluded epileptic myoclonic seizures but was suggestive of nonepileptic faciobrachial dystonic seizures. In these circumstances, CSF and EEG investigations are very important in the differential diagnosis, and include a negative LGI1 antibody, positive 14-3-3 brain protein, and PSWCs on the EEG.
In summary, focal PSWCs on the EEG, faciobrachial dystonic seizures, and hyponatremia may occasionally be found in patients with CJD. The frequency of such findings is not known, with only few cases reported in the literature. Hence clinicians should not be dissuaded from a diagnosis of CJD when faciobrachial dystonic seizures and hyponatremia are found.
Conclusion
Our case showed atypical manifestations and nonspecific EEG findings of CJD, which may be confused with anti-LGI1 encephalitis. Thus, the presence of faciobrachial dystonic seizures and hyponatremia in a patient with progressive dementia and psychiatric symptoms may suggest a number of neurological diseases, but CJD needs to be considered. The pathophysiology also needs further study.
Acknowledgment
We thank Jiang Wu who helped to develop the manuscript and revised it critically for important intellectual content.
Author contributions
LS participated in drafting of the manuscript. JC collected the clinical data. CL participated in the study design and coordination and helped draft the manuscript. YL was responsible for the design and agreed to be accountable for the integrity of all aspects of the work. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Disclosure
The authors report that there are no conflicts of interest with respect to the research, authorship, funding, and/or publication of this article.
References
Du Plessis DG, Larner AJ. Phenotypic similarities causing clinical misdiagnosis of pathologically confirmed sporadic Creutzfeldt-Jakob disease as dementia with Lewy bodies. Clin Neurol Neurosurg. 2008;110:194–197. | ||
Haïk S, Brandel JP, Sazdovitch V, et al. Dementia with Lewy bodies in a neuropathologic series of suspected Creutzfeldt-Jakob disease. Neurology. 2000;55:1401–1404. | ||
Demaerel P, Baert AL, Vanopdenbosch L, Robberecht W, Dom R. Diffusion-weighted magnetic resonance imaging in Creutzfeldt-Jakob disease. Lancet. 1997;349:847–848. | ||
Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913–920. | ||
Zerr I, Bodemer M, Gefeller O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol. 1998;43:32–40. | ||
Steinhoff BJ, Racker S, Herrendorf G, et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:162–166. | ||
World Health Organization. Consensus on criteria for diagnosis of sporadic CJD. Wkly Epidemiol Rec. 1998;73:361–365. | ||
Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55:811–815. | ||
Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jacob disease. Ann Neurol. 2004;56:702–708. | ||
Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.