")
Back to Journals » OncoTargets and Therapy » Volume 8
Cell death in response to antimetabolites directed at ribonucleotide reductase and thymidylate synthase
Authors Asuncion Valenzuela MM, Castro I, Gonda A, Diaz Osterman C, Jutzy J, Aspe J, Khan S , Neidigh J, Wall N
Received 20 December 2014
Accepted for publication 21 January 2015
Published 26 February 2015 Volume 2015:8 Pages 495—507
DOI https://doi.org/10.2147/OTT.S79647
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Malyn M Asuncion Valenzuela, Imilce Castro, Amber Gonda, Carlos J Diaz Osterman, Jessica M Jutzy, Jonathan R Aspe, Salma Khan, Jonathan W Neidigh, Nathan R Wall
Center for Health Disparities and Molecular Medicine, Division of Biochemistry, Department of Basic Sciences, Loma Linda University, Loma Linda, CA, USA
Abstract: New agent development, mechanistic understanding, and combinatorial partnerships with known and novel modalities continue to be important in the study of pancreatic cancer and its improved treatment. In this study, known antimetabolite drugs such as gemcitabine (ribonucleotide reductase inhibitor) and 5-fluorouracil (thymidylate synthase inhibitor) were compared with novel members of these two drug families in the treatment of a chemoresistant pancreatic cancer cell line PANC-1. Cellular survival data, along with protein and messenger ribonucleic acid expression for survivin, XIAP, cIAP1, and cIAP2, were compared from both the cell cytoplasm and from exosomes after single modality treatment. While all antimetabolite drugs killed PANC-1 cells in a time- and dose-dependent manner, neither family significantly altered the cytosolic protein level of the four inhibitors of apoptosis (IAPs) investigated. Survivin, XIAP, cIAP1, and cIAP2 were found localized to exosomes where no significant difference in expression was recorded. This inability for significant and long-lasting expression may be a reason why pancreatic cancer lacks responsiveness to these and other cancer-killing agents. Continued investigation is required to determine the responsibilities of these IAPs in their role in chemoresistance in pancreatic adenocarcinoma.
Keywords: IAPs, exosomes, pancreatic cancer, antimetabolites, gemcitabine, cladribine, hydroxyurea, 5-fluorodeoxyuridine, 5-fluorouracil
Introduction
Pancreatic adenocarcinoma is the fourth leading cause of cancer death.1,2 Out of all diagnosed patients, only 2%–5% survive 5 years, and the average survival time is only 4–6 months.3–6 There are a number of treatments available for patients, but the option for metastatic pancreatic cancer is limited to chemotherapy, of which only the antimetabolite drugs gemcitabine (Gem) and 5-fluorouracil (5FU) are clinically used.3,7,8 Antimetabolite drugs are designed to stop deoxyribonucleic acid (DNA) synthesis and replication. Each antimetabolite differs from one another in their mechanism of actions, leading to different cytotoxic effects. 5FU is converted into its active form FdUMP, which then acts as a thymidine synthase inhibitor. It inhibits the conversion of dUMP to dTMP by binding to thymidine synthase and folate.9,10 Unlike 5FU, Gem has three mechanisms of action, making this antimetabolite a broad-spectrum agent. Once Gem enters the cell, it is subjected to phosphorylation by deoxycytidine kinase into Gem monophosphate. Conversion of Gem into its di- and triphosphorylation states by nucleoside kinases converts this antimetabolite into a ribonucleotide reductase and polymerase inhibitor, respectively.11–13 There have been numerous efforts to improve chemotherapy treatment regimens by combining these chemotherapies with either 5FU or Gem in combination. Unfortunately, most of these studies have confirmed that combination therapy does not show significant improvements.14–16 In addition to the failure to improve the treatment regimen, patients face the challenge of chemoresistance. A low response rate in patients treated with Gem has been shown to be associated with innate and acquired chemoresistance.17 Additional studies still need to be conducted to understand resistance to Gem and 5FU in pancreatic cancer patients.
The inhibitor of apoptosis (IAP) family of proteins includes survivin, XIAP, cIAP1, and cIAP2. IAPs are characterized by a ~70 amino acid baculovirus IAP repeat (BIR) domain and, except for survivin, a RING domain in the C-terminus of each family member.18,19 XIAP directly binds to activated caspase-3, -7, and -9 using its BIR domains, inhibiting the caspases’ function.20–23 On the other hand, while cIAP1 and cIAP2 are weak caspase inhibitors,24 these IAPs act as E3 ubiquitin–protein isopeptide ligases on Smac using their RING domains to promote Smac degradation.25 Survivin, the smallest IAP, is both structurally and functionally unique among the rest of the IAP family, having a multifunctional role in various cellular activities, which includes the regulation of mitosis, protection from cell death, and adaptation to stressful environments.26–28 This IAP is also found to be localized in the cytoplasm, mitochondria, and nucleus, with its subcellular location determining its function.29,30 Our lab has shown that an extracellular pool of survivin exists, which causes neighboring cancer cells to become resistant to therapy, to rapidly proliferate, and to acquire an increased potential to be invasive.31
Recently, our lab has discovered that survivin is released by small (40–100 nm) membrane-bound vesicles called exosomes.32 Tumor cell-derived exosomes (TEX) have been shown to be released constitutively into the extracellular space,33 both in vitro and in vivo.34,35 TEX have different molecular profiles and biological roles, giving an indication of the cell of origin.36,37 In addition, specific protein content found on and within TEX and exosomes in general establishes their functional role.38 The goal of this study is to examine whether antimetabolite treatments in PANC-1 cells modulate IAP protein and message levels both intracellularly and exosomally. Such IAP modulation may indicate that these chemotherapeutic agents may contribute to chemoresistance in pancreatic cancer cells.
Materials and methods
Cell cultures
The pancreatic carcinoma (PANC-1) cell line was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (ATCC) supplemented with 100 U penicillin, 100 μg/mL streptomycin, 100 μg/mL normocin (Invitrogen, Grand Island, NY, USA), and 10% fetal bovine serum (CellGro, Manassas, VA, USA). The cells were grown in a humidified atmosphere of 37°C in 95% O2/5% CO2 until 60% confluent. Their conditioned medium (CM) for exosome collection was collected after 24 hours of treatment with cladribine (CldA), Gem, hydroxyurea (HU), 5-fluorodeoxyuridine (5FdU), and 5FU (Sigma-Aldrich Co., St Louis, MO, USA). All antimetabolites were dissolved in water and various concentrations were added to cells. For CM collection for exosome isolation, cells were plated 24 hours prior to treatment. Medium was changed before antimetabolite treatment to ensure no apoptotic bodies were present. PANC-1 cells were treated for 24 hours, after which CM and cells for Western blots and polymerase chain reaction (PCR) were harvested.
Apoptosis and cell proliferation analysis
Cells at 60% confluency and 37°C were treated with vehicle (water) or various doses of antimetabolites for 24, 48, and 72 hours. Cells were harvested and stained with annexin V and propidium iodide (BioLegend, San Diego, CA, USA) per the manufacturer’s directions. Apoptosis and cell proliferation studies were performed and analyzed using an MACS Quant flow cytometer and FlowJo software (Tree Star, Ashland, OR, USA).
Exosome isolation
Exosomes were isolated as previously described39 with the following modifications. Briefly, CM was centrifuged three times prior to ultracentrifugation: 400× g for 10 minutes to remove cells, 2,000× g for 20 minutes to remove cell debris, and 10,000× g for 30 minutes to remove nucleic acid and soluble albumin. The supernatant was collected and stored at −80°C until needed. Exosomes were isolated from the CM by ultracentrifugation on a 30% sucrose cushion at 100,000× g for 16 hours. The exosomes in the sucrose cushion were extracted and washed once in PBS by ultracentrifugation at 100,000× g for 2 hours. The exosome pellet was resuspended in 100 μL PBS or lysis buffer.
Exosomes were also isolated using ExoQuick TC™ (Systems Biosciences, Mountain View, CA, USA). Briefly, CM was collected from the treated cells and centrifuged at 3,000× g for 15 minutes. Two milliliters of ExoQuick TC™ was mixed to 10 mL of CM and incubated at 4°C overnight. Following incubation, the CM was centrifuged at 1,500× g for 30 minutes to pellet exosomes. The exosome pellet was resuspended in the appropriate buffer and used for ribonucleic acid (RNA) extraction studies.
Exosome quantification
The amount of exosomes released was semiquantified by assessing the acetylcholinesterase activity, as our lab has described previously.39 Briefly, 40 μL of the isolated exosomes was suspended in 110 μL PBS. The PBS-diluted exosome fraction was equally divided to three individual wells on a 96-well flat-bottomed microplate. Acetylcholine (1.25 mM) and 5,5′-dithiobis(2-nitrobenzoic acid) (0.1 mM) were added to the exosomes to a total volume of 300 μL. The change in absorbance at 412 nm was monitored every 5 minutes for 30 minutes.
To determine total exosome number, exosomes were diluted 1:100 in PBS from the total isolated exosome sample and analyzed using a NanoSight LM10-HS microscope (Wiltshire, UK). Size distribution and total number of exosomes per milliliter were calculated by the nanoparticle tracking analysis software (Wiltshire, UK).
Western blots
For total cell Western blot analysis, cells are harvested and lysed in cell lysis buffer (0.5% Triton X-100, 300 mM NaCl, 50 mM Tris/HCl, 1 mM PMSF) with sonication. The lysates were centrifuged at 10,000 rpm at 4°C for 20 minutes to remove cell debris. For exosome Western blot analysis, exosomes were solubilized in lysis buffer. Protein concentration was determined using the BCA protein assay (Pierce Chemical, Rockford, IL, USA). A total of 50 μg cellular protein or 40 μg exosomal protein was separated using 7.5%–12% sodium dodecyl sulfate polyacrylamide gels and transferred onto nitrocellulose membrane (Bio-Rad Laboratories Inc., Hercules, CA, USA). Blots were immunostained with antibodies against survivin (1:500–2,000, Novus Biologicals, Littleton, CO, USA), cIAP1, cIAP2, and XIAP (1:500–1,000, Cell Signaling, Danvers, MA, USA). β-actin (1:1,000, Cell Signaling) was used as control for cell samples, and Lamp-1 (1:500, BioLegend, San Diego, CA, USA) was used as control for exosome samples. Goat antirabbit and antimouse antibodies (LI-COR Biosciences, Lincoln, NE, USA) were used as secondary antibody. The immunoreactive bands were visualized using the Odyssey imaging system (LI-COR Biosciences).
PCR
Harvested cells and isolated exosomes were resuspended in TRI Reagent® (Molecular Research Center, Cincinnati, OH, USA) and stored at −80°C until needed. RNA was extracted per manufacturer’s directions. RNA quantification was performed using a NanoDrop 2000c (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription of RNA was performed using the First Strand cDNA Synthesis Kit (Syd Labs, Inc., Malden, MA, USA). Genomic DNA was eliminated prior to reverse transcription of RNA into cDNA. A total concentration of 100 ng/μL cDNA was utilized to perform PCR reactions using Phusion® Flash High-Fidelity PCR Master Mix (Finnzymes, Thermo Scientific, Pittsburgh, PA, USA). Forward and reverse primers (IDT, San Diego, CA, USA) have been designed to detect survivin, cIAP1, cIAP2, and XIAP genes (Table 1).
 | Table 1 Forward and reverse primer dimers for inhibitor of apoptosis (IAP) analysis |
cDNA was amplified for detection of survivin, cIAP1, cIAP2, XIAP, and GAPDH (housekeeping gene) using the LightCycler 1.0 system real-time thermal cycler and the LightCycler FastStart DNA MasterPLUS SYBR Green kit (Roche Applied Science). The assessment of samples as positive for expression was based on 1) observing reproducible cycle threshold (CT) values in two replicates of the target gene where the GAPDH housekeeping gene showed a CT value of <30 cycles and 2) melting curve analysis showing superimposable melting curves across samples and replicates for a given target gene in the absence of a superimposable product in negative control samples. Gene-specific primer pairs were designed to span introns using Roche’s Assay Design Center (www.roche-applied-science.com). Primers were as follows: GAPDH forward, 5′-GAG TCC ACT GGC GTC TTC AC; GAPDH reverse, 5′-GTT CAC ACC CAT GAC GAA CA; survivin forward, 5′-ATG GGT GCC CCG ACG TT; survivin reverse, 5′-TCA ATC CAT GGC AGC CAG; XIAP forward, 5′-GAC AGT ATG CAA GAT GAG TCA; XIAP reverse, 5′-GCA AAG CTT CTC CTC TTG CAG; cIAP1 forward, 5′-AGC TAG TCT GGG ATC CAC CTC; cIAP1 reverse, 5′-GGG GTT AGT CCT CGA TGA AG; cIAP2 forward, 5′-TGG AAG CTA CCT CTC AGC CTA C; cIAP2 reverse, 5′-GGA ACT TCT CAT CAA GGC AGA. Cycling parameters for all products were initial denaturation of 15 minutes at 95°C followed by 50 cycles of 10 seconds at 95°C (denaturing), 5 seconds at 60°C (annealing), and 15 seconds at 72°C (elongation).
Statistical analysis
Statistical analysis was performed using a two-way analysis of variance with the aid of GraphPad Prism statistical software (La Jolla, CA, USA), with paired t-test used for group analysis. Densitometric analysis was conducted using our Licor Odyssey Imager (Licor, Lincoln, NE, USA). Density of individual bands was divided by β-actin, GAPDH, or LAMP-1 as the internal controls for cytosolic cellular protein, block PCR mRNA samples, or exosomal proteins, respectively, with each sample then divided by the particular baseline control.
Results
Antimetabolite treatments induce growth inhibition and cell death in PANC-1 cells
CldA treatment of PANC-1 cells with various doses (50 nM, 100 nM, 1 μM, 20 μM) showed that lower doses did not inhibit cell growth or show cytotoxic effects (Figure 1A and B). At 1 μM CldA, there was a decrease in cell proliferation but no significant killing, unlike treatment at 20 μM. Treatment with Gem (1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM) showed a time- and dose-dependent killing effect, while growth inhibition was evident at all time points and doses except for 1 nM (Figure 1C and D). Increasing the concentration of HU (5 μM, 50 μM, 100 μM, 500 μM, 1 mM) reduced cell proliferation in a time- and dose-dependent manner. However, the drug’s cytotoxic effects were only evident with the two highest doses (Figure 1E and F). Treatment with 5FdU (100 pM, 1 nM, 10 nM, 100 nM, 1 μM, 10 μM) (Figure 1G and H) and 5FU (100 nM, 500 nM, 1 μM, 5 μM, 50 μM) showed similar cytotoxic and growth inhibition profiles (Figure 1I and J). Of interest, the killing effects of the drugs were time and dose dependent, while cell proliferation was only reduced by the higher doses at all time points (data not shown).
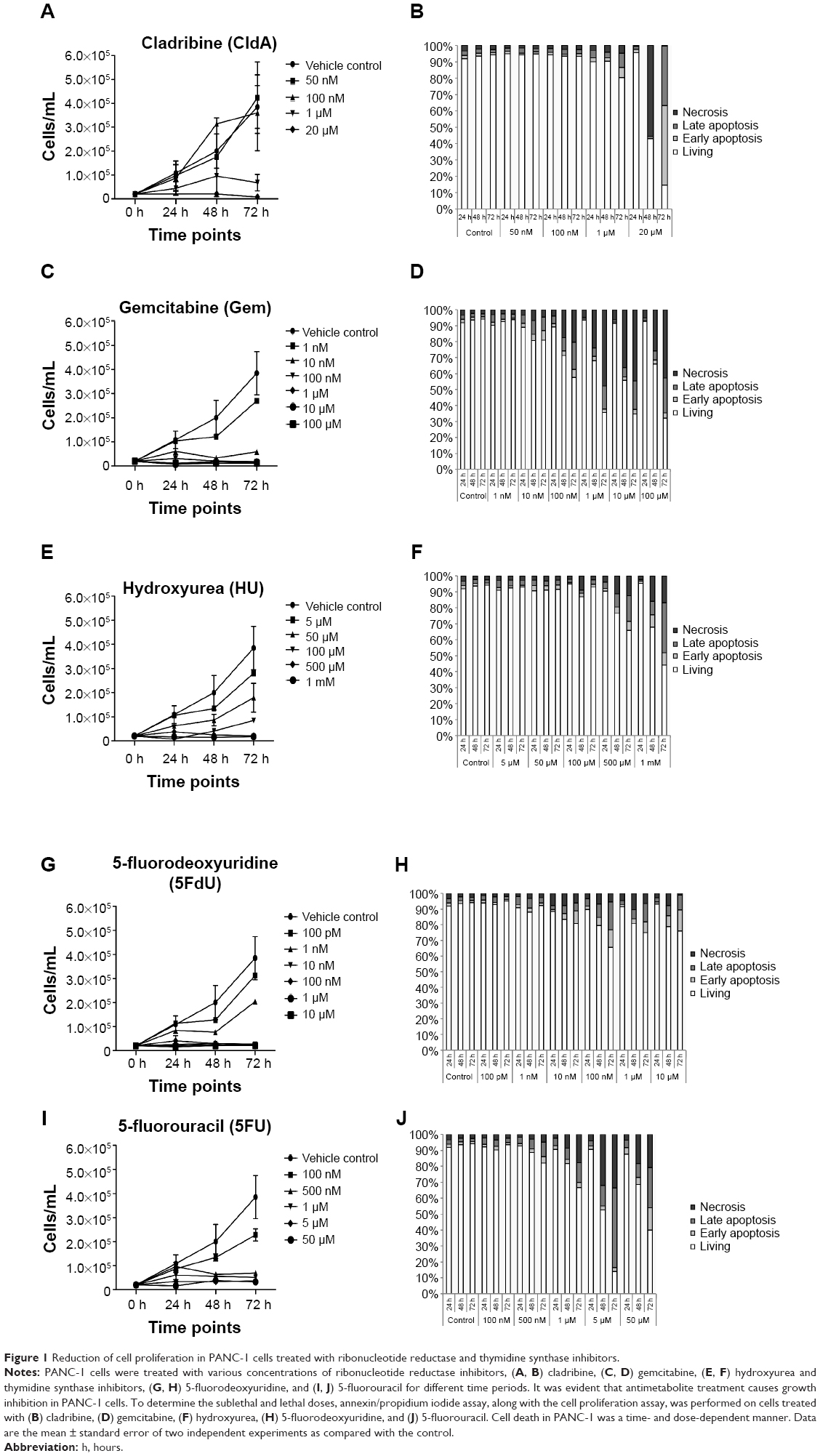 | Figure 1 Reduction of cell proliferation in PANC-1 cells treated with ribonucleotide reductase and thymidine synthase inhibitors. |
IAPs expression levels are not reduced by antimetabolite treatments and do not play a role in inhibiting cell death in PANC-1 cells
To determine whether IAPs play a part in cell death inhibition in PANC-1 cells and play a role in chemoresistance, sublethal and lethal doses were chosen to stress the cells for 24, 48, and 72 hours. Ribonucleotide reductase inhibitors did not significantly alter the protein expression of IAPs at 24 and 48 hours (Figure 2A and B). Although not significant, modulation of survivin expression was shown after 72 hours, in particular with HU treatment at 500 μM (Figure 2C). In both mRNA (Figures 2D and S1) and protein, IAP expression levels were either maintained or increased across the doses of ribonucleotide reductase inhibitors.
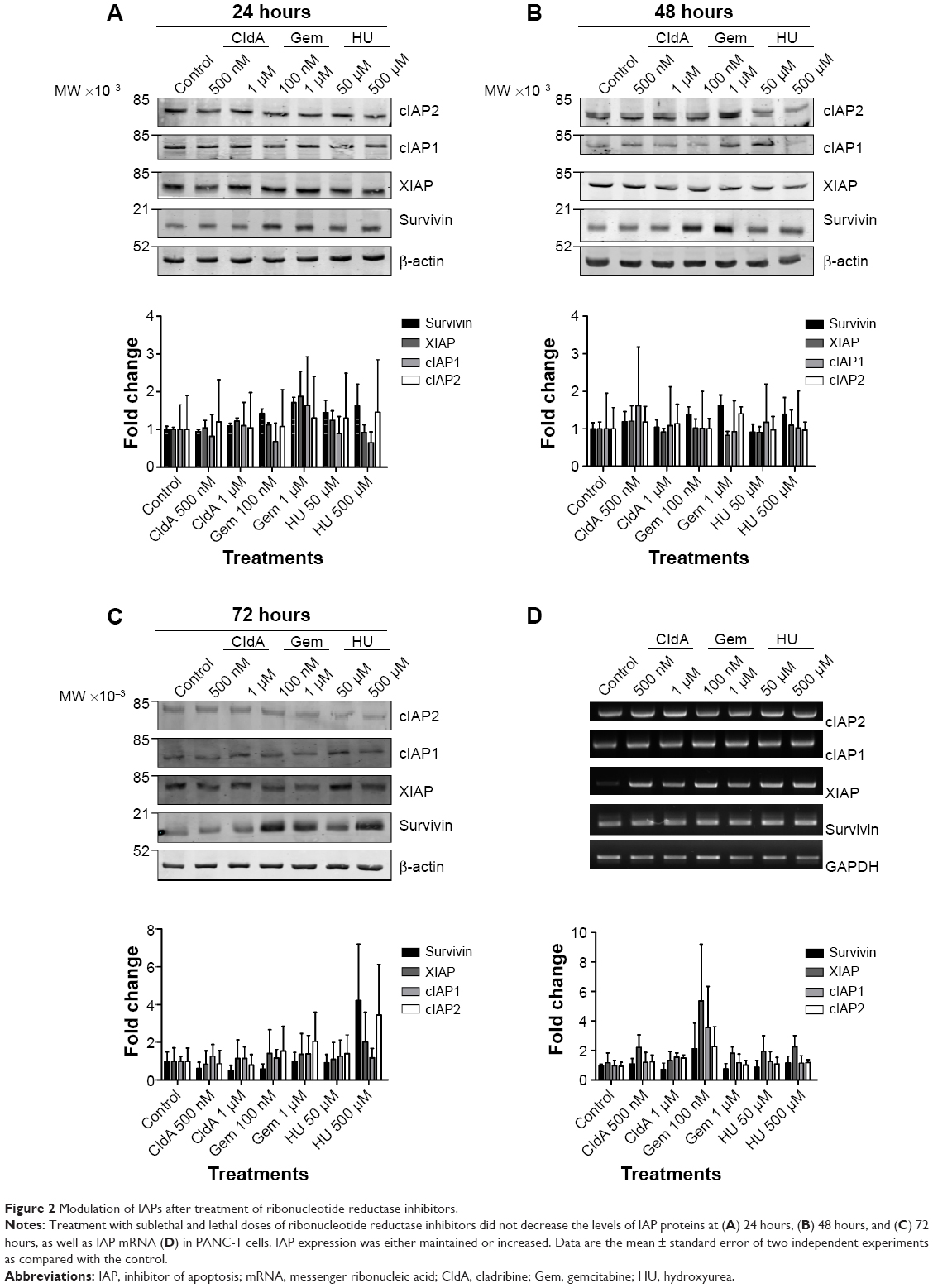 | Figure 2 Modulation of IAPs after treatment of ribonucleotide reductase inhibitors. |
As shown with the ribonucleotide reductase inhibitors, treatments with thymidine synthase inhibitors did not significantly alter IAP protein (Figure 3A–C) or mRNA (Figures 3D and S1) expression levels across all time points and doses. Following the same trend as with the other antimetabolite family, the IAP protein and mRNA were either maintained or modestly increased.
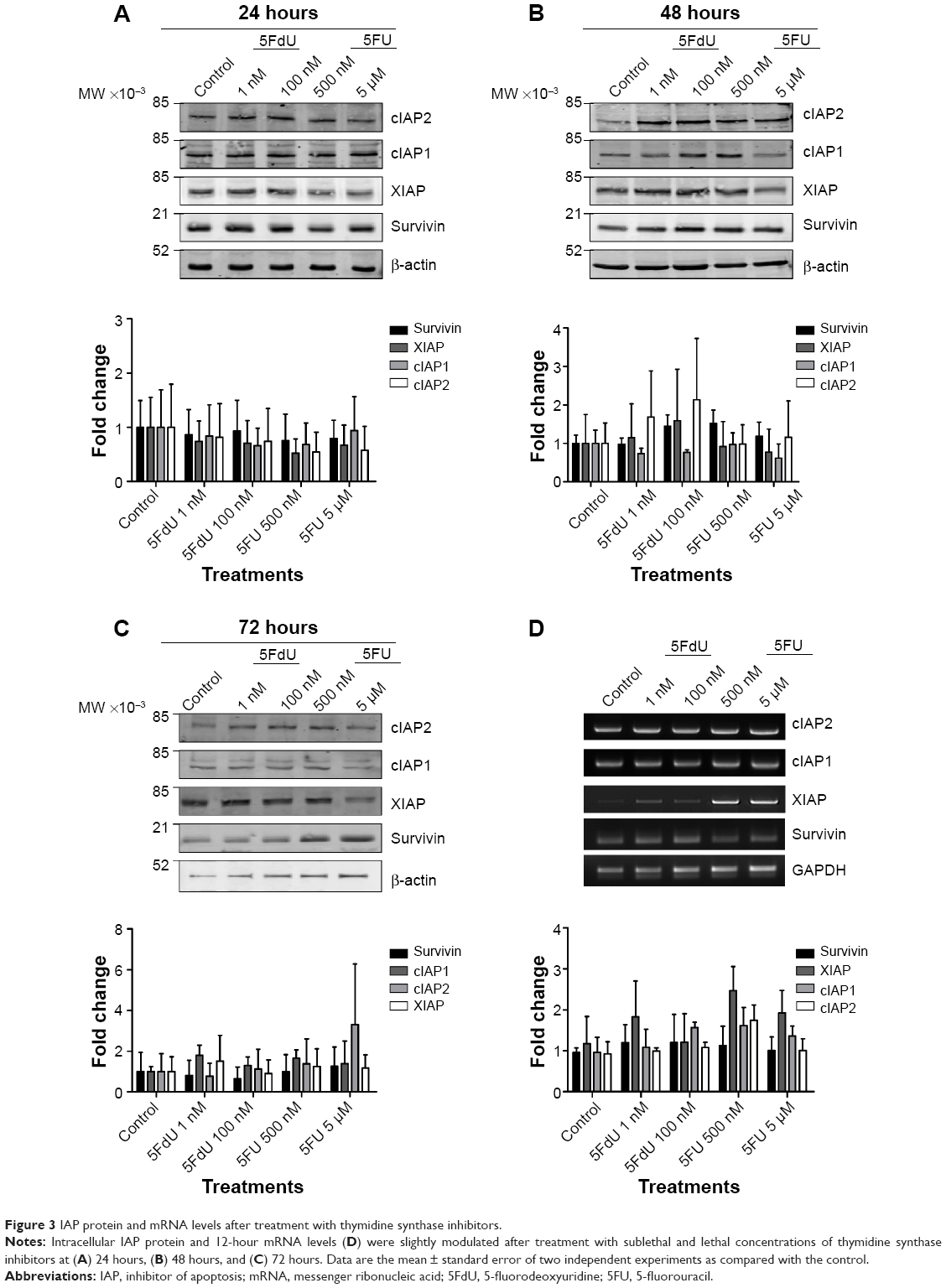 | Figure 3 IAP protein and mRNA levels after treatment with thymidine synthase inhibitors. |
Exosome amount released changes with treatment
To determine whether antimetabolite drug treatment stress would affect the amount of exosomes released, an AChE assay was performed. There was no significant difference in the amount of exosomes released in the untreated cells compared with the treated cells (Figure 4A). Interestingly, the BCA protein assay showed less total protein concentration in the untreated cell sample compared with the treated cells (Figure 4B). To verify the results, exosomes were examined using a NanoSight LM10-HS, which determines the number of exosomes present per milliliter. Figure 4C shows that compared with the untreated exosome sample, there were more exosomes present in the pooled sample of treated exosomes.
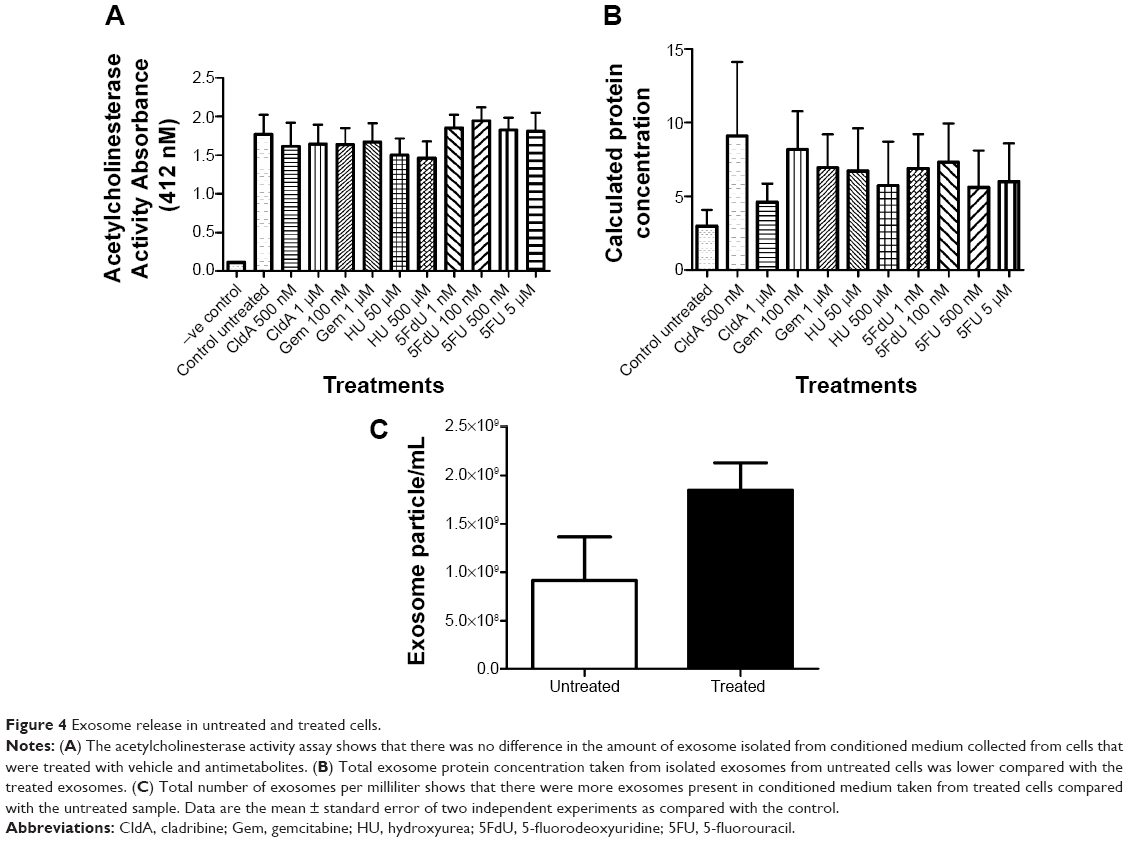 | Figure 4 Exosome release in untreated and treated cells. |
Protein and mRNA inhibitor of apoptosis proteins released via exosomes
To further investigate whether IAPs, in addition to survivin, are released into the extracellular space, exosomes were isolated from CM taken from treated and nontreated cells after 24 hours. Western blotting was performed to determine the presence of IAP proteins in exosomes. As shown in Figure 5A and B, not only was survivin present in exosomes but so were XIAP, cIAP1, and cIAP2. In addition, treatment with antimetabolites affected the levels of some released exosomal IAP proteins. Treatment with ribonucleotide reductase inhibitors (Figure 5A) decreased the levels of exosomal cIAP2, cIAP1, XIAP, and survivin at all concentrations, while exosomal IAPs released from cells treated with thymidine synthase inhibitors were only effective in reducing the exosomal levels of cIAP1, cIAP2, and XIAP (Figure 5B). Though survivin was modestly reduced after the treatment of 5FdU, 5FU was unable to change its level (Figure 5B).
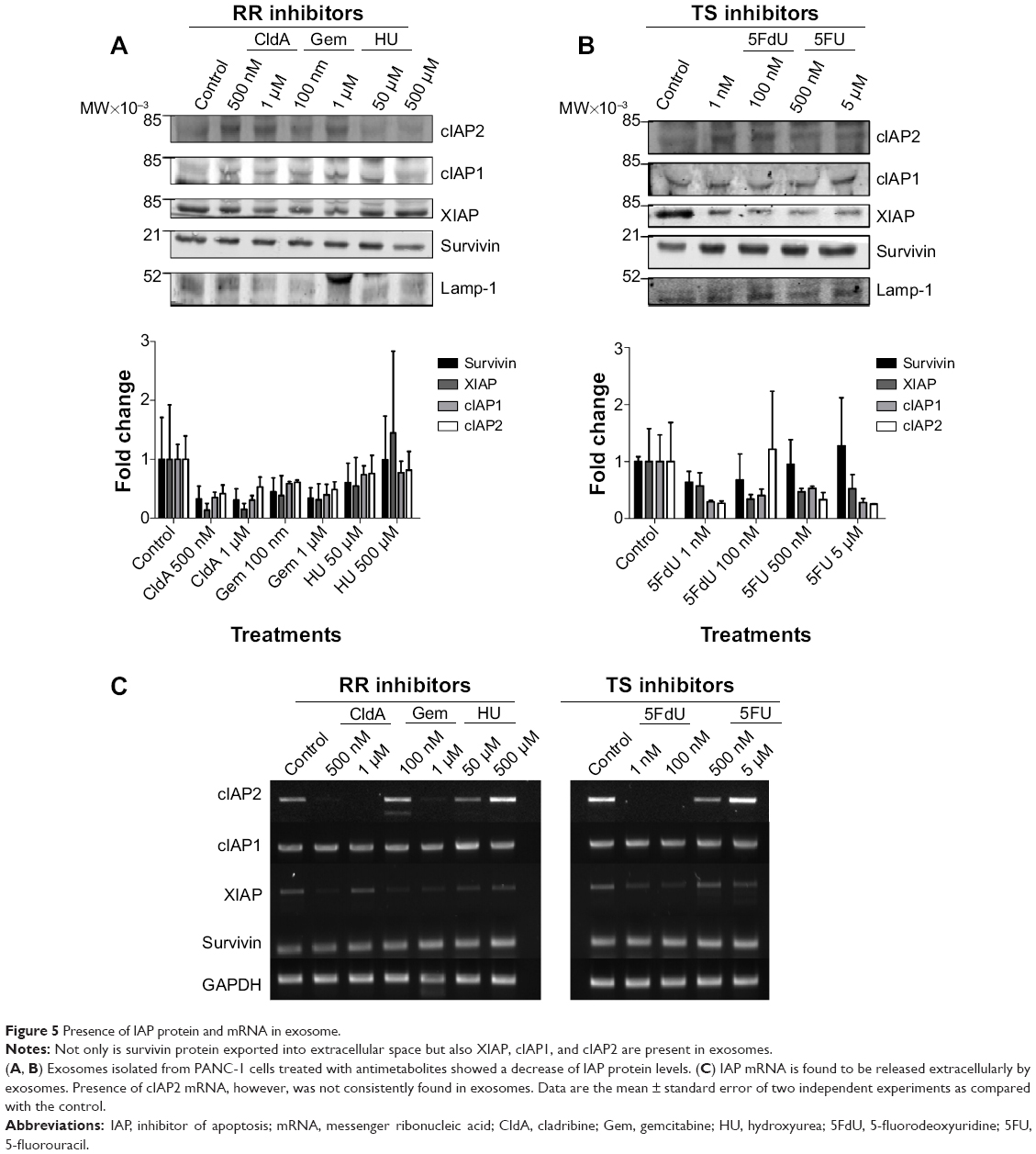 | Figure 5 Presence of IAP protein and mRNA in exosome. |
We next determined whether IAP mRNAs were also present in exosomes. PCR analysis indicated that all IAPs were present in exosomes (Figures 5C and S1B). Surprisingly, while survivin and cIAP1 mRNA levels remained unchanged with treatment, the presence of cIAP2 and XIAP mRNA was not consistent across the samples (Figures 5C and S1B). In addition, XIAP mRNA levels decreased in the majority of the treatments using both families of inhibitors.
Discussion
In various types of cancer, the function and/or expression of the IAPs is not properly regulated. This can be due to a decrease in levels of endogenous IAP inhibitors, abnormalities in the gene, or an increase in the expression of either mRNA or protein.40 Specifically, the high levels of survivin expression in cancer cells have been associated with dismal prognosis, disease progression, metastatic dissemination, chemo- and radiotherapy resistance, and overall dismal disease outcome.28,41,42 In many cancer types, such as glioblastoma, renal cell carcinoma, and liver and pancreatic cancer, the chromosome region of 11q21–23, which includes both the cIAP1 and cIAP2 genes, is shown to be amplified in these diseases, making cIAP1/2 protooncogenes. Additionally, in about 50% of surveyed mucosa-associated lymphoid tissue (MALT) lymphoma cases, the BIR domain of cIAP2 is fused to the C-terminus of the paracaspase mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1). This fusion protein in turn constitutively activates NF-κB.40,43 Overexpression of XIAP correlated with poor clinical outcome, lower survival rates, and aggressive tumor growth in diffuse large B-lymphoma, colorectal cancer, and clear-cell renal cell carcinoma, respectively.40 In pancreatic adenocarcinoma specifically, it has been shown that survivin, XIAP, cIAP1, and cIAP2 are constitutively upregulated by NF-κB in cell lines and tissue samples. This abnormal upregulation of IAPs also correlates with chemotherapy resistance.44 Numerous efforts have been made to target these IAPs to address the problem with resistance to therapy. XIAP silencing by siRNA in pancreatic cancer cell lines has been shown to increase Gem sensitivity,45 as well as an enhancement of cell death when treated with both Gem and proton radiation.42 Other studies have targeted survivin, showing that knocking this IAP down with siRNA caused Gem chemosensitivity.46 Chemotherapy treatment for metastatic pancreatic cancer is limited, and modulation of IAP protein and message levels by these antimetabolites is yet to be determined, both intracellularly and exosomally.
In this study, we first determined that CldA, Gem, and HU were all able to decrease cell proliferation in addition to inducing cell death in a time- and dose-dependent manner (Figure 1A–F). We expected IAP protein and mRNA levels to be modulated by the ribonucleotide reductase inhibitors in both sublethal and lethal doses, as cell death was evident at these doses. Surprisingly, we observed that the IAP levels were maintained or even increased (Figure 2). The sublethal dose of Gem was not able to decrease the levels of IAP mRNA, but instead increased expression of all four IAPs. In addition, the lethal dose of Gem consistently increased the levels of IAP protein across all time points.
Like the ribonucleotide reductase inhibitors, cell proliferation was decreased with treatments of 5FdU and 5FU. Cell death was also evident in a time- and dose-dependent manner (Figure 1G–J). However, treatments with the sublethal and lethal doses also did not reduce IAP protein and mRNA expression levels (Figure 3). Since cell death was shown in both sublethal and lethal doses, we expected the IAP protein and mRNA levels to be modulated by these agents. Surprisingly, we observed that the IAP levels were maintained or even increased. This indicates that cell death shown in Figure 1 may not result from a decrease of IAP levels in these cells but is through a different mechanism. It is thus not surprising that studies using antimetabolite compounds against leukemias have been recently shown to overcome apoptosis resistance and trigger necroptotic cell death.47 Additionally, the failure to reduce the levels of IAPs intracellularly by not only Gem and 5FU but all the other agents in both antimetabolite families may play a role in chemoresistance in pancreatic adenocarcinoma patients and why combination therapies do not improve patient survival rates. In studies involving colon cancer cells, cIAP2 reduction has proven to be the only means to increase the efficacy of 5FU.48,49
Tumor exosomes have been described as “multipurpose carriers”, having a supportive role in the survival and growth of the tumor cells and being involved in promoting host tissue invasion and the subsequent metastasis and facilitating immune response evasion.50–52 It is speculated that the diverse function of tumor exosomes is due to the various bioactive molecules on and within the vesicles, which strongly influences the tumor microenvironment.53–56 We have also shown that extracellular survivin has a significant effect on the tumor microenvironment, causing cells to become highly proliferative, invasive, and resistant to therapy.31 In addition to survivin being exosomal, we found that XIAP, cIAP1, and cIAP2 proteins and mRNA are also released into the extracellular space via exosomes (Figure 5).
We looked at the exosomal levels of IAP proteins treated with ribonucleotide reductase inhibitors and observed that the intracellular IAP expression levels did not reflect the levels of extracellular IAP expression (Figure 5A). Here, the levels of exosomal IAPs were reduced, with the exception of the treatment of HU (500 μM). Looking at the exosomal IAP levels, we saw that 5FU treatments increased survivin levels in comparison with the other IAPs, as with the cIAP2 with 5FdU treatment at 100 nM (Figure 5B). We expected the exosomal IAP levels would reflect the IAP levels found intracellularly. However, there were modest reductions in the exosomal IAP protein levels in the treated samples. We hypothesize that in an attempt to compensate for decreasing levels of IAPs in the exosomes, chemotherapy-treated cells released more exosomes into the extracellular space (Figures 4 and 5). IAP mRNAs were also present in the exosomes (Figures 5C and S1B). The levels of cIAP1 and survivin appear rather consistent across treatment, but the inconsistent presence of XIAP and cIAP2 mRNA may be due to truncated mRNA that is found within the exosomes. Further work in our lab and others is still evaluating this possibility.
Taken together, the results of this study suggest that protein and mRNA IAPs are found in exosomes and that both cellular and exosomal IAPs should be investigated for their roles in drug resistance in pancreatic cancer. Moreover, though these antimetabolites reduced survival and cell proliferation, levels of the four IAPs studied here only modestly changed and at times increased in both locations depending upon the IAP. Our findings demonstrate for the first time that IAP protein and mRNAs are found in exosomes. More studies, however, are needed in order to fully determine the function of exosomal IAPs in the extracellular space and whether they exhibit similar effects as extracellular survivin. We also showed that other players are most likely involved in the cell death of PANC-1 cells after antimetabolite treatments, while the failure to decrease the levels of both protein and mRNA intracellular IAPs may play a role in chemoresistance in pancreatic cancer patients. Although Gem was not able to reduce intracellular IAP protein and mRNA levels, it continues to be the first-line treatment against metastatic pancreatic cancer. The ability to have different mechanisms of action depending on its phosphorylation state may be why Gem continues to be superior compared with other antimetabolites.57 The failure to significantly decrease the levels of both protein and mRNA intracellular IAPs may play a role in chemoresistance in pancreatic cancer patients. Innate and acquired chemoresistance in patients is a continuing problem in the clinic. Therefore, it is important to continue to find better ways to treat pancreatic adenocarcinoma to try to overcome the problem of resistance and improve overall patient survival rates.
Acknowledgments
This work would have been impossible if not for a generous grant from the Hirshberg Foundation for Pancreatic Cancer Research and the friendship, inspiration, and mentoring of Agi Hirshberg. Funding for our laboratory has also come from grants for health disparity research: NIH-NCMHD Project EXPORT Program 5P20MD001632/Project 3 (NRW). Funding also came from a National Merit Test Bed (NMTB) award sponsored by the Department of the Army under Cooperative Agreement Number DAMD17-97-2-7016 (NRW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank the entire NRW lab for careful review of the manuscript.
Disclosure
Dr Nathan Wall is the interim CEO of Canget BioTekpharma LLC for which he receives travel reimbursements for travel associated with this venture. All other authors declare no conflict of interest.
References
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. | ||
Niederhuber JE, Brennan MF, Menck HR. The National Cancer Data Base report on pancreatic cancer. Cancer. 1995;76:1671–1677. | ||
Warshaw AL, Fernandez-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326:455–465. | ||
Coleman MP, Gatta G, Verdecchia A, et al. EUROCARE-3 summary: cancer survival in Europe at the end of the 20th century. Ann Oncol. 2003;14:128–149. | ||
Carpelan-Holmstrom M, Nordling S, Pukkala E, et al. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data Finnish Cancer Registry. Gut. 2005;54:385–387. | ||
Wagner M, Redaelli C, Lietz M, Seiler CA, Friess H, Buchler MW. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br J Surg. 2004;91:586–594. | ||
Ahrendt SA, Pitt HA. Surgical management of pancreatic cancer. Oncoogy. 2002;16:725–734. | ||
Moss RA, Lee C. Current and emerging therapies for the treatment of pancreatic cancer. Onco Targets Ther. 2010;3:111–127. | ||
Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. | ||
Thomas DM, Zalcberg JR. 5-Fluorouracil: a pharmacological paradigm in the use of cytotoxics. Clin Exp Pharmacol Physiol. 1998;25:887–895. | ||
Andersson R, Aho U, Nilsson BI, et al. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44:782–786. | ||
Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17:7–12. | ||
Giovannetti E, Mey V, Danesi R, Mosca I, Del Tacca M. Synergistic cytotoxicity and pharmacogenetics of gemcitabine and pemetrexed combination in pancreatic cancer cell line. Clin Cancer Res. 2004;10:2936–2943. | ||
Poplin E, Feng Y, Berlin J, et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2009;27(23):3778–3785. | ||
Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23(15):3509–3516. | ||
Louvet C, Andre T, Lledo G, et al. Gemcitabine combined with oxaliplatin in advanced pancreatic adenocarcinoma: final results of a GERCOR multicenter phase II study. J Clin Oncol. 2002;20:1512–1518. | ||
Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320(2):138–149. | ||
Srinivasula SM, Ashwell JD. IAPs: what’s in a name? Mol Cell. 2008;30:123–135. | ||
Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. 2010;430:199–205. | ||
Deveraux QL, Reed JC. IAP family proteins-suppressors of apoptosis. Genes Dev. 1999;13:239–252. | ||
Miller LK. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 1999;9:323–328. | ||
Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. | ||
Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. | ||
Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281(6):3254–3260. | ||
Hu S, Yang X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J Biol Chem. 2003;278(12):10055–10060. | ||
Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struc Biol. 2000;7:602–608. | ||
Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. | ||
Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. | ||
Li F. Survivin study: what is the next wave? J Cell Physiol. 2003;197:8–29. | ||
Li F, Ackermann EJ, Bennett CF, et al. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. | ||
Khan S, Aspe JR, Asumen MG, et al. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br J Cancer. 2009;100:1073–1086. | ||
Khan S, Jutzy JMS, Aspe JR, McGregor DW, Neidigh JW, Wall NR. Survivin is released from cancer cells via exosomes. Apoptosis. 2011;16(1):1–12. | ||
Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. | ||
Ginestra A, La Placa MD, Saladino F, Cassara D, Nagase H, Vittorelli ML. The amount of proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res. 1998;18(5A):3433–3437. | ||
Ginestra A, Miceli D, Dolo V, Romano FM, Vittorelli ML. Membrane vesicles in ovarian cancer fluids: a new potential marker. Anticancer Res. 1999;19(4C):3439–3445. | ||
Andre F, Schartz NE, Movassagh M, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360(9329):295–305. | ||
Wieckowski E, Whiteside TL. Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles. Immunol Res. 2006;36(1–3):247–254. | ||
Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600. | ||
Khan S, Jutzy JMS, Aspe JR, et al. The application of membrane vesicles for cancer therapy. Advances in Cancer Therapy: Intech. 2011:21–52. 2011. | ||
Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–124. | ||
Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulators of mitosis and apoptosis and novel targets for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. | ||
Galloway NR, Aspe JR, Sellers C, Wall NR. Enhanced antitumor effect of combined gemcitabine and proton radiation in the treatment of pancreatic cancer. Pancreas. 2009;38(7):782–790. | ||
Lau R, Pratt MA. The opposing roles of cellular inhibitor of apoptosis proteins in cancer. ISRN Oncol. 2012;2012:928120. | ||
Lopes RB, Gangeswaran R, McNeish IA, Wang Y, Lemoine NR. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer. 2007;120(11):2344–2352. | ||
Shrikhande SV, Kleeff J, Kayed H, et al. Silencing of X-linked inhibitor of apoptosis (XIAP) decreases gemcitabine resistance of pancreatic cancer cells. Anticancer Res. 2006;26(5A):3265–3273. | ||
Yoon DH, Shin JS, Jin DH, et al. The survivin suppressant YM155 potentiates chemosensitivity to gemcitabine in the human pancreatic cancer cell line MiaPaCa-2. Anticancer Res. 2012;32(5):1681–1688. | ||
Chromik J, Safferthal C, Serve H, Fulda S. Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett. 2014;344(1):101–109. | ||
Miura K, Karasawa H, Sasaki I. cIAP2 as a therapeutic target in colorectal cancer and other malignancies. Expert Opin Ther Targets. 2009;13(11):1333–1345. | ||
Karasawa H, Miura K, Fujibuchi W, et al. Down-regulation of cIAP2 enhances 5-FU sensitivity through the apoptotic pathway in human colon cancer cells. Cancer Sci. 2009;100(5):903–913. | ||
Nieuwland R, Sturk A. Why do cells release vesicles? Thromb Res. 2010;125. | ||
Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007;67(7):2912–2915. | ||
Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lan Invest. 2010;90(11):1549–1557. | ||
van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006;140(1):13–21. | ||
Mears R, Craven RA, Hanrahan S, et al. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics. 2004;4(12):4019–4031. | ||
Hegmans JP, Bard MP, Hemmes A, et al. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol. 2004;164(5):1807–1815. | ||
Iero M, Valenti R, huber V, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15(1):80–88. | ||
Valenzuela MMA, Neidigh JW, Wall NR. Antimetabolite treatment for Pancreatic Cancer. Chemotherapy. 2014;3(3):1–7. |
Supplementary material
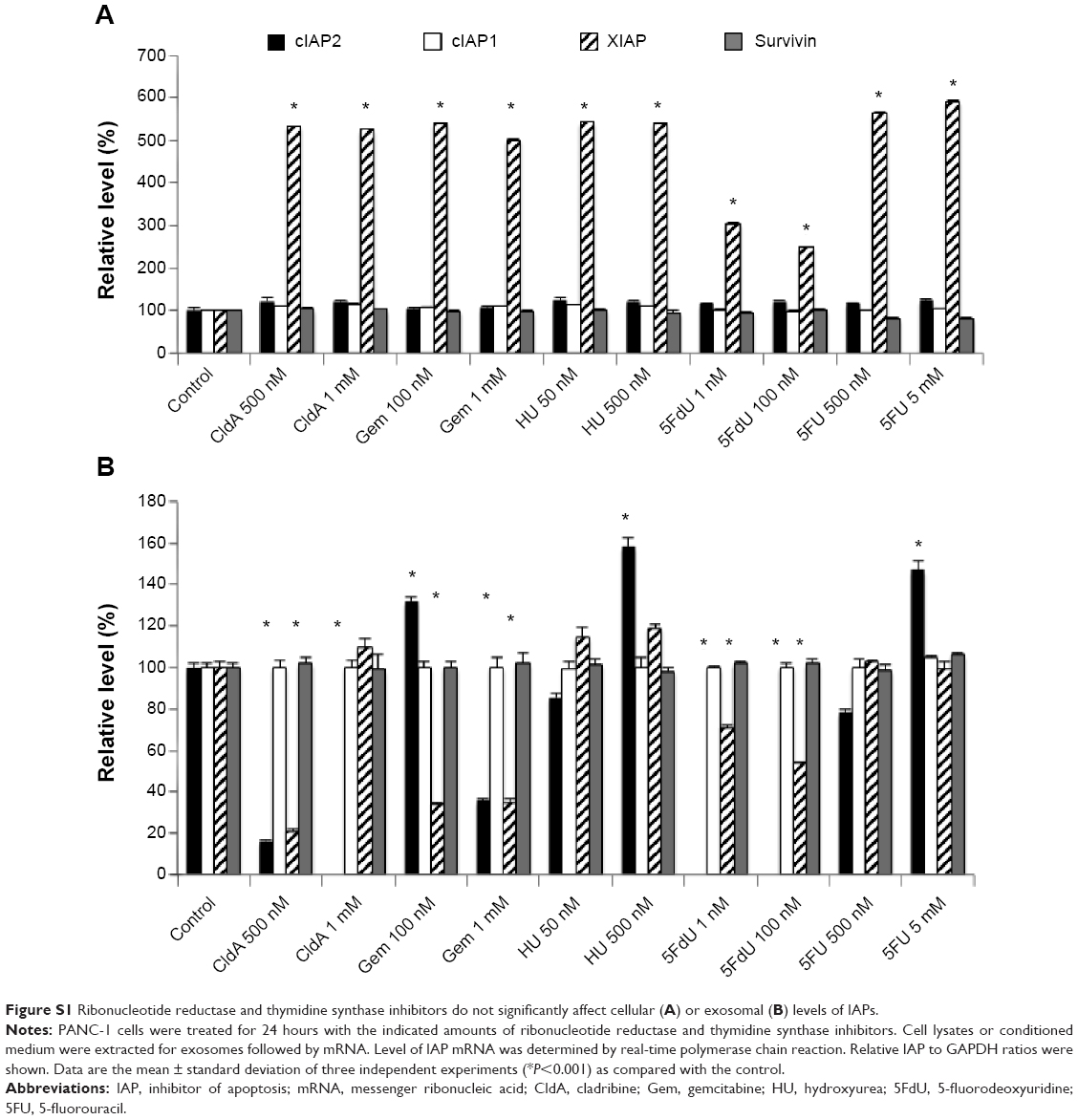 | Figure S1 Ribonucleotide reductase and thymidine synthase inhibitors do not significantly affect cellular (A) or exosomal (B) levels of IAPs. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.