")
Back to Journals » Transplant Research and Risk Management » Volume 7
Cardiac allograft immune activation: current perspectives
Authors Chang D, Kobashigawa JA
Received 12 July 2014
Accepted for publication 26 August 2014
Published 18 December 2014 Volume 2015:7 Pages 13—22
DOI https://doi.org/10.2147/TRRM.S71055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Qing Yi
Video abstract presented by David Chang and Jon Kobashigawa.
Views: 347
David Chang, Jon Kobashigawa
Cedars-Sinai Heart Institute, Los Angeles, CA, USA
Abstract: Heart transplant remains the most durable option for end-stage heart disease. Cardiac allograft immune activation and heart transplant rejection remain among the main complications limiting graft and recipient survival. Mediators of the immune system can cause different forms of rejection post-heart transplant. Types of heart transplant rejection include hyperacute rejection, cellular rejection, antibody-mediated rejection, and chronic rejection. In this review, we will summarize the innate and adaptive immune responses which influence the post-heart transplant recipient. Different forms of rejection and their clinical presentation, detection, and immune monitoring will be discussed. Treatment of heart transplant rejection will be examined. We will discuss potential treatment strategies for preventing rejection post-transplant in immunologically high-risk patients with antibody sensitization.
Keywords: heart transplant, innate immunity, adaptive immunity, rejection, immunosuppression
Introduction
The burden of heart failure continues to grow, with an excess of 5 million patients in the United States with this clinical syndrome.1 End-stage, or stage D, heart failure affects approximately 5% of this population, in which 1-year mortality can be as high as 75% with medical therapy alone.2 Options for stage D heart failure are limited, but include guideline-directed medical therapy, palliative care (including chronic inotropic therapy), mechanical circulatory support, heart transplant, and experimental therapies, such as stem cell trials. Heart transplantation remains the most durable treatment for end-stage heart disease that is not amenable to other treatment modalities, such as anti-ischemic or antiarrhythmic therapies. In a recent report from the International Society for Heart and Lung Transplantation (ISHLT), the median survival after heart transplant is 11 years.3 According to this report, however, less than 2,500 heart transplants surgeries are completed each year in North America. Thus, donor availability for heart transplant is far below the need from potential heart transplant recipients. For this reason, left ventricular assist devices and cardiac replacement therapies, such as the Total Artificial Heart (SynCardia Systems Inc., Tucson, AZ, USA), can be used as a bridge to heart transplant. These devices are being utilized with increasing frequency prior to heart transplant as approximately 33% of heart transplant recipients had a mechanical device as a bridge to transplant. Left ventricular assist devices and Total Artificial Heart therapies offer promise to patients who need mechanical support prior to heart transplant. Complications after placement of mechanical circulatory support include infection and blood transfusion. These factors, among others, can lead to antibody production known as antibody sensitization.4 Antibody formation to major histocompatibility complex (MHC) class I and class II human leukocyte antigens (HLAs) place an individual at higher immunologic risk post-heart transplant.
Mechanisms of immune responses
The innate and adaptive immune systems work in conjunction to ward off pathologic infection. In dysregulation, these systems can also cause autoimmune disease in non-immunosuppressed patients or cause rejection in solid organ transplant recipients. While there is significant interaction between the innate and adaptive immune systems, to date, modulation of the adaptive immune system has been the focus of chronic immunosuppression post-transplant. The role of the innate immune system post-transplant is a topic of current interest in translational research.
Components of the innate immune system include leukocytes (neutrophils, eosinophils, and mastocytes), macrophages, dendritic cells, and natural killer (NK) cells.5 Cells of the innate immune system interact against a defined set of molecules including pathogen-associated molecular patterns in the context of infection and damage-associated molecular patterns in the process of tissue injury. Toll-like receptors (TLRs) are one type of pattern-recognition receptor (PRR) involved in initiation and amplification of innate immune responses.6 Cells of the innate immune system, including dendritic cells, can recognize TLRs. Different TLRs are expressed on different cell surface types (endothelial or epithelial cells) or in intracellular compartments. TLRs assist in early detection of microbes, but can also recognize markers of cell damage, including heat shock proteins7 and nucleic acids released by necrotic cells. TLR activation is amplified through signaling, primarily through myeloid differentiation factor 88 (MyD88).8 Recognition of TLR by dendritic cells aids in the maturation of the dendritic cell, allowing dendritic cell interactions with effector T lymphocytes. In this manner, stimulation of the innate immune system can, in turn, activate the adaptive immune system. Cytokines and chemokines are released in response to activation of the innate immune system via TLR signaling.9 Chemokine release can, in turn, recruit mediators of the adaptive immune response, including leukocytes, to sites of tissue injury. At the time of heart transplant, reperfusion injury and tissue injury can activate such components of the innate immune system to stimulate cytokine release to activate the complement cascade leading to further cell injury and cell death. The complement cascade has been implicated in acute and chronic rejection processes. The C5a and C3a components of the complement cascade may assist in T-cell co-stimulation.10,11 NK cells impact the innate immune response by recognizing foreign cells in the absence of antigen-specific interactions. In this manner, NK cells can directly kill donor cells. Antigens from deceased donor cells can be processed by antigen-presenting cells (APCs), leading to indirect stimulation of the adaptive immune system by the NK cell.
Primary mediators in the adaptive immune system include T lymphocytes and B lymphocytes. T lymphocytes develop in the thymus through processes of positive and negative selection. T-cells can be subdivided into cytotoxic T-cells (CD8+ T-cells) and CD4+ T-helper cells. The CD4+ T-helper cells can function as effector or regulatory T-cells. Differential cytokine milieu can influence differing CD4 T-cell phenotypes.12 Interferon (IFN)-gamma and interleukin (IL)-12 promote Th1 cells that are important in host defense mechanisms against intracellular pathogens. IL-4 drives TH2 CD4 T-cells that combat extracellular parasites. IL-6 and transforming growth factor (TGF)-beta lead to TH17 cells that combat yeast, fungi, and extracellular bacteria. TH17 cells can also influence autoimmune diseases including multiple sclerosis, psoriasis, rheumatoid arthritis, and Crohn’s disease. Tfh cells, or T follicular helper cells, aid germinal center B-cells in plasma cell differentiation. B lymphocytes develop in the spleen and lymph nodes. B-cells are the primary antibody-producing cells of the adaptive immune system. Both T- and B-cells can proliferate in response to activation and form a memory response. With repeat activation, the response is augmented and amplified. T-cells interact with B-cells and other APCs (including macrophage and dendritic cell lines of the innate immune system) via the MHC class II cell surface receptor on the APC and the T-cell receptor (TCR) of the T-cell. MHC I antigens are constitutively expressed on a majority of cells and help delineate self versus non-self (non-self or foreign protein/s that would elicit an immune response). TCR engagement of class I or class II MHC leads to T-cell activation. Co-stimulation via B7-CD28 and CD40-CD40L interactions between APCs and T-cells are also required for T-cell activation. These activated T-cells are a main mediator of the adaptive immune response and contribute to heart transplant rejection. In fact, most standard chronic immunosuppression post-heart transplant is directed at suppressing T-cell activation via suppressing T-cell signaling or T-cell proliferation.
Activated effector T- and B-cells are pathologic to the transplanted graft. Regulatory T-cells may serve to downregulate an activated immune system and antagonize the process of allograft immune activation. There are numerous types of regulatory cells. Regulatory T-cells include CD4+ T-cells that express regulatory transcription factor forkhead box (Fox) p3+ (Tregs), CD8+Foxp3+ Tregs, CD4–CD8– T-cells, and NK T-cells.13 Tregs in humans can express Foxp3, Fas receptor (CD95), and cytotoxic T-lymphocyte antigen 4 (CTLA4). These Tregs are of considerable interest as they can exert potent immunoregulatory effects, including APC killing, consumption of IL-2, and downregulation of the immune system. Degranulation of mastocytes can lead to loss of Tregs and can contribute to heart transplant rejection.14
Heart transplant rejection
Post-heart transplant, mediators of innate and adaptive immunity influence the possible development of rejection. The risk of rejection is highest in the first year post-heart transplant.15 Risk of rejection in the first year post-transplant has decreased from 33% of recipients in 2004 to 25% of recipients in 2010. Moreover, rejection requiring treatment has decreased from 25% in 2004 to 14% in 2010.3 Hyperacute rejection, acute cellular rejection (ACR), acute antibody-mediated (humoral) rejection, and chronic rejection can affect the functional status and longevity of the heart transplant recipient. The main goal of immunosuppression is to prevent heart transplant rejection. Further understanding of the mediators of rejection will aid in prevention and treatment of these disease processes.
Hyperacute rejection can occur immediately post-heart transplant and is manifest in critical cardiogenic shock and profound organ hypoperfusion. Diagnosis is made usually on clinical suspicion, and mechanical support with extracorporeal membrane oxygenation is often required along with aggressive antirejection therapies. This process is difficult to distinguish from severe primary graft dysfunction. Hyperacute rejection is thought to be an immunological process caused by preformed recipient antibodies to the graft. It has been reported in ABO blood type incompatibility as well.16 Hyperacute rejection is currently a rare clinical scenario due to the development of the prospective cytotoxic crossmatch17,18 and, more recently, use of the virtual crossmatch.19,20 The prospective cytotoxic crossmatch, or complement-dependent, cytotoxicity-based assay, combines recipient serum (potential source of donor-specific antibodies [DSA]) with donor lymphocytes. Cytotoxicity with addition of exogenous complement represents a positive crossmatch and significant DSA against donor class I and/or class II HLA. The prospective crossmatch is still used in select highly sensitized recipients, but, more frequently, the virtual crossmatch is utilized, as the prospective crossmatch requires local expertise and is time-consuming. The virtual crossmatch utilizes information from solid-phase assays that allow detection of the specificity and binding strength of an antibody.21 With use of these assays, the immunologic MHC class I and II status of the recipient and donor can be virtually matched for compatibility. Prior to transplant, the recipient’s probability of potential donor compatibility can be calculated against a panel of reactive antibodies.22 Calculated panel reactive antibody (cPRA) data are based on allele frequencies from HLA phenotypes of deceased kidney donors. While there are differing definitions of antibody sensitization pre-transplant, a patient with a cPRA >10% may be considered sensitized. Highly sensitized individuals have cPRA >50% and may require desensitization treatment prior to transplant.23 A novel assay to detect the potential functional significance of antibodies has been described. This C1q assay detects a subset of IgG antibodies that are capable of fixing complement. The C1q assay was useful in prediction of C1q+ DSA contributing to antibody mediated rejection (AMR) episodes and graft loss following kidney transplant.24
ACR is primarily a T-cell-mediated process with graft infiltration by leukocytes and macrophages. ACR requiring treatment may present insidiously. It may also manifest with new-onset symptomatic heart failure, with rhythm disturbances, or with cardiogenic shock. The most common rhythm disorder seen in acute rejection is atrial fibrillation. Bradycardia in the setting of heart transplant rejection is an ominous finding. The diagnosis of cellular rejection is definitively made by endomyocardial biopsy.25 Sampling error may lead to false negative biopsy results. Endomyocardial biopsy, however, remains the gold standard in the diagnosis of heart transplant rejection. Grading of cellular rejection is based on standardized guidelines (see Table 1).26 In this grading scale, grade 2 R and 3 R cellular rejections generally require treatment. Decrement in left ventricular systolic function by echocardiogram is highly suggestive of rejection as well. Subtherapeutic immunosuppressant trough levels predispose recipients to cellular rejection. Inadequate immunosuppression may be due to medication noncompliance, nonadherence to medications due to side effect or cost, poor medication absorption, infection, or drug interactions. Risk factors for ACR include a higher number of HLA mismatches, age of recipient, female sex, race, and use of induction therapy.27 Young patients are at higher risk of rejection, and African-American recipients may have a genetic predisposition to require higher doses of effective immunosuppressant therapy, possibly due to polymorphisms in cytochrome P450 enzymes used for drug metabolism. Survival rates are reduced in patients who require treatment for cellular rejection.
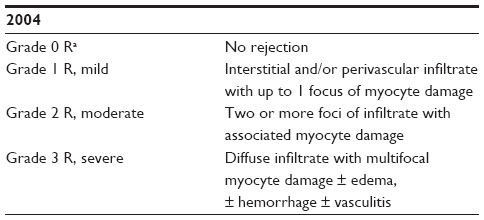 | Table 1 ISHLT standardized cardiac biopsy grading: acute cellular rejection |
In comparison to decreased incidence of ACR, AMR is increasingly recognized, likely in part due to antibody sensitization in transplant recipients. AMR was first described in 1987;28 further consensus of the clinical importance and definition of AMR was published in 2011.29,30 AMR occurs in approximately 15% of heart transplant recipients. There is a wide range of clinical presentations in patients with AMR, from asymptomatic patients to patients with hemodynamic compromise and cardiogenic shock. In AMR, the vascular endothelium may be the primary antigenic stimulus. Antibodies to activated endothelial cells may interact with the complement cascade and lead to adverse remodeling with macrophage infiltration and complement deposition. Donor graft-specific MHC class I or class II antibodies may be present in the graft or in the serum, but are not requisite for the diagnosis of AMR, as DSA may be absent in the serum if they are tissue-bound. When present in the serum post-transplant, whether transient or persistent, de novo donor HLA-specific antibodies portend a poor prognosis.31 Diagnosis of AMR is based on histology and immunohistochemistry (Table 2). Histologic changes include myocardial capillary injury, endothelial swelling, intravascular macrophage accumulation, intravascular thrombus, and myocyte necrosis. Immunohistochemical evidence of AMR includes positive stains directed against macrophages (CD68), endothelium (CD31, CD34), or complement products (C4d). AMR has been associated with hemodynamic compromised rejection, cardiac allograft vasculopathy (CAV), and increased mortality.32–34 Asymptomatic AMR also predicts poor outcomes post-heart transplant.35 Risk factors for AMR include female sex, elevated cPRA pre-transplant, positive complement-dependent cytotoxicity or virtual crossmatch, Cytomegalovirus seropositivity, pre-transplant mechanical assist device, and retransplantation.15
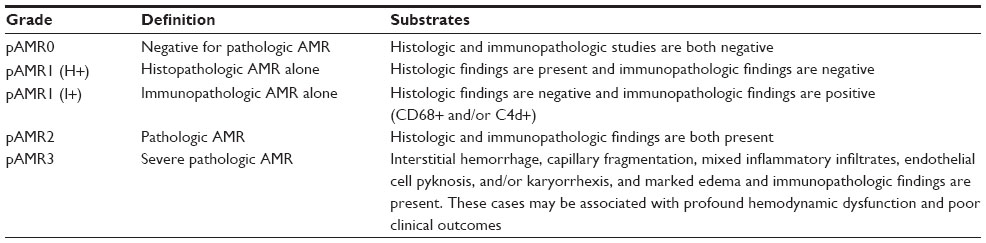 | Table 2 The 2013 ISHLT working formulation for pathologic diagnosis of cardiac antibody-mediated rejection |
Chronic rejection, also known as CAV, has an association with AMR. CAV remains a leading cause of morbidity and mortality post-heart transplant. CAV is reported to impact 50% of heart transplant recipients within 10 years after transplant. CAV, in contrast to native coronary disease, is generally thought to be diffuse panarteritis with diffuse, concentric intimal thickening of the coronary arteries. A recent consensus statement from the ISHLT helped further define CAV (Table 3).36 Change in maximal intimal thickness >0.5 mm between early intravascular ultrasound (IVUS) (6–8 weeks) and at 1 year post-transplant has been shown to be predictive of CAV-related mortality and nonfatal major adverse coronary events.37 Immune and nonimmune factors contribute to development of CAV. Nonimmune factors include contributions from hypertension, hyperlipidemia, and oxidant stress.38 In the denervated state post-heart transplant, patients with CAV are unlikely to present with angina. Routine angiography should be considered to evaluate for presence and severity of CAV. Conventional coronary angiography remains the gold standard with respect to assessment of coronary vasculature. Alternatives to the invasive coronary angiogram include exercise-based or pharmacologic stress testing, cardiac positron emission tomography, and coronary computed tomography angiography.39–42 Additional modalities, such as optical coherence tomography, may augment conventional coronary angiographic and IVUS data in the evaluation of CAV.43
 | Table 3 Recommended nomenclature for cardiac allograft vasculopathy |
Detection of rejection and immune monitoring
The gold standard for diagnosis of ACR or AMR remains the assessment of the endomyocardial biopsy. Cardiac magnetic resonance imaging has shown promise in diagnosis of rejection.44 This modality may be particularly useful in cases of biopsy-negative rejection. As clinical and echocardiographic manifestations may present late, many transplant centers will have a protocol-based schedule for endomyocardial biopsy with right heart catheterization to routinely evaluate for ACR, AMR, and cardiac hemodynamics. Biopsies are most frequent in the first month post-transplant and taper gradually in time. When rejection is present and treated, our institutional practice involves follow-up biopsy within 2 weeks to ensure that the findings on biopsy improve or normalize after treatment. Routine assessment post-biopsy with echocardiography will give further information regarding cardiac function and structure. Data are limited regarding accurate diagnosis of heart transplant rejection by noninvasive echocardiographic-based methods.45
To reduce patient discomfort and anxiety due to the invasive nature of endomyocardial biopsy, there are alternative tests that can be performed to assess for heart transplant rejection. The most commonly used noninvasive blood test used to assess the possibility of ACR in low-risk individuals is the AlloMap® (CareDx, Inc., Brisbane, CA, USA). This test uses gene expression profiling and was first studied between 6 months and 5 years post-transplant in patients at low risk for rejection.46,47 The test has a reported excellent negative predictive value, but the positive predictive value is low. Abnormal AlloMap studies prompt a safety endomyocardial biopsy to further assess for cellular rejection. Use of the AlloMap in the first year post-transplant has also been examined.48 There are a number of limitations of the AlloMap . Its use has been studied in a low-risk patient population in single-organ (heart) transplant.47 It is important to note that the AlloMap does not assess AMR. Hemodynamics from a right heart catheterization, which would routinely follow an endomyocardial biopsy, are not obtained. Finally, an abnormal AlloMap would prompt a safety endomyocardial biopsy. Despite its limitations, in appropriately selecting patients, the AlloMap test does have clinical utility in the assessment of cellular rejection.49
Another noninvasive assay with clinical utility to monitor levels of immunosuppression is the Cylex test (ImmuKnow®; Cylex Inc., Columbia, MD, USA). The Cylex test assesses adenosine triphosphate production from phytohemagglutinin (PHA)-stimulated T-cells. Low Cylex values can suggest over-immunosuppression and risk of infection.50 Elevated values of the Cylex score in theory may reflect under-immunosuppression and risk of rejection. Elevated Cylex values have been reported to correlate with increased plaque progression by IVUS even at 2 months post-transplant, which suggests that elevated Cylex scores could potentially signal risk of AMR, given the association of AMR with CAV.51
Chronic maintenance immunosuppression and treatment of rejection
Chronic maintenance immunosuppressant therapy post-heart transplant generally involves therapies directed to suppress the T-cell. These therapies include a combination of calcineurin inhibition (CNI) with tacrolimus (Prograf®; Astellas Pharma Inc., Tokyo, Japan) or cyclosporine (Gengraf®; Abbvie Inc., North Chicago, IL, USA) to inhibit signaling downstream of the TCR; purine antagonism with mycophenolate mofetil or azathioprine to attenuate T-cell proliferation; and pan-immunosuppressant steroid therapy. Ideally, patients can be weaned off prednisone in the first year post-heart transplant should they maintain normal cardiac function, be free of rejection, and not have development of DSA. CNI monotherapy has been used to attempt to minimize the adverse sequelae of immunosuppression.52,53 With monotherapy-based immunosuppression, target trough levels may be targeted in a higher range, which may lead to differences in the side effect profiles observed. For example, in CNI monotherapy, higher target trough levels could, in theory, lead to renal insufficiency. In combination immunosuppressive therapy, should a patient have an episode of rejection, one could consider transition from purine antagonist to proliferation signal inhibition (PSI) with sirolimus (Rapamune®; Wyeth Pharmaceuticals Inc., Philadelphia, PA, USA) or everolimus (Zortress®; Novartis International AG, Basel, Switzerland). This transition may reduce the risk of rejection given the results of a clinical trial comparing three arms of immunosuppressant therapies.54 A recent trial reported success with early PSI use and CNI withdrawal within 3 months post-heart transplant55 The risk/benefit profile of purine antagonist versus PSI therapy must be weighed on a case-by-case basis, as some clinical trials for immunosuppression with PSI have shown possible deleterious effects on renal function and risk of infection.56,57
Treatment of rejection depends on patient symptoms and the severity of the rejection episode. With treatment for rejection, there is always a balance between augmented immunosuppression with potential for improvement in graft function and the risk of infection. Treatment for AMR in asymptomatic patients with preserved cardiac function is controversial, as its benefit has not been proven. ACR can be treated with oral or intravenous (IV) steroid bolus and taper and CNI target levels can be increased. For patients experiencing ACR and AMR with reduced cardiac function or with clinical symptoms, pulse-dose IV steroids, rabbit anti-thymocyte globulin (ATG), and/or IV immunoglobulin (IVIG) can be utilized. ATG is a cytolytic antibody that binds the CD3 moiety associated with the TCR, leading to T-cell depletion. The mechanism of IVIG is not fully understood, but is likely pleomorphic.58 IVIG can affect immune activation by neutralization of pathologic antibodies via anti-idiotypic interactions, competition for fragment, crystallizable portion of an antibody (Fc) binding sites, and inhibition of complement activity. Patients that present with heart failure or cardiogenic shock may have hyperacute rejection immediately post-transplant, delayed hyperacute rejection within 1 week post-transplant, or, later, a combination of ACR and AMR. These patients are at high risk for significant morbidity and death and are treated with IV pulse steroids as a pan-immunosuppressant, plasmapheresis to remove pathologic antibodies, cytolytic therapy with ATG to deplete T-cells, and IVIG. There are limited data supporting the use of B-cell-targeted therapies in acute AMR.59 Treatment of acute rejection with bortezomib (Velcade®; Millenium Pharmaceuticals, Inc., Cambridge, MA, USA) in conjunction with plasmapheresis is another option that has not been studied in detail in heart transplant recipients, but has shown benefit in renal transplant patients with ACR and AMR.60 Bortezomib is a proteasome inhibitor that selectively depletes antibody-producing plasma cells. Patients that are critically ill due to rejection require close hemodynamic monitoring and may require directed inotropic therapy, mechanical support with an intra-aortic balloon pump, or potentially, extracorporeal membrane oxygenation support.61 With aggressive therapy, there is hope that the immune activation process will be downregulated and that cardiac function will normalize (Table 4).
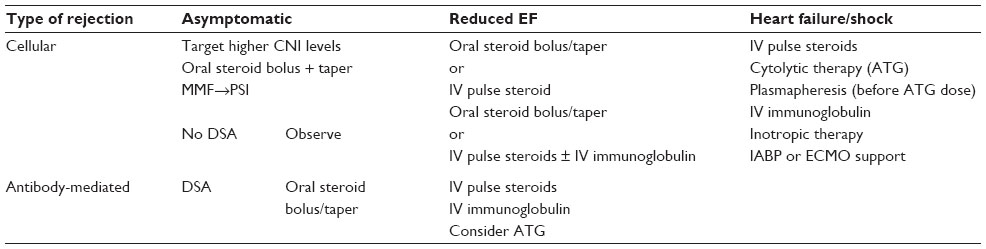 | Table 4 Treatment modalities for cellular and/or humoral rejection |
After treatment for rejection, patients who develop DSA may benefit from further treatment with IVIG and rituximab (Rituxan®; Genentech Inc., South San Francisco, CA, USA), a humanized mouse monoclonal antibody that depletes CD20+ B-cells. One such approach would use IVIG at 1 g/kg daily for 2 days, followed by rituximab therapy commenced at 1 week (1 g or 375 mg/m2 if the patient is <50 kg). Repeat IVIG dosing can be commenced at 1 month. These therapies need to be temporally separated, as IVIG may bind to and attenuate the efficacy of rituximab. Serial flow cytometric HLA should be assessed 2 weeks after treatment. After treatment for acute rejection, for patients that have recurrent or persistent rejection on endomyocardial biopsy, additional intense immunosuppressant treatment may be warranted. Further treatments of recurrent or persistent rejection include total lymphoid radiation and photopheresis. Total lymphoid radiation is not often used due to concerns for future development of malignancy. Photopheresis has been used for immunomodulation. The process involves treatment of a minority (~5%) of peripheral lymphocytes with 8-methoxypsoralen and ultraviolet light, inducing T-cell apoptosis. This process of T-cell depletion may have an impact on Tregs. Photopheresis, in contrast to other modalities of antirejection treatment, may not increase infectious risk. Patients with rejection are not candidates for repeat heart transplant within 6 months of an episode of rejection as the immunologic milieu of the patient may acutely reject an organ upon retransplantation. Rarely, durable mechanical assist devices are used as a bridge to recovery or retransplantation
Treatment of immunologically high-risk sensitized patients prior to and after heart transplant
While immunosuppressant therapies are required post-heart transplant to prevent or treat cardiac allograft activation, the use of immunomodulating therapies prior to heart transplant is under investigation. There is evidence that supports desensitization strategies prior to renal transplant with IVIG and rituximab.62 Prior to heart transplant, patients with elevated cPRA may benefit from desensitization therapies to potentially reduce the risk of post-transplant rejection and expand the number of compatible donor candidates available for heart transplant. As described above for patients who develop DSA post-transplant, IVIG and rituximab can be used in a desensitization protocol prior to heart transplant. For patients that do not reduce their antibody burden with IVIG and rituximab therapy, a pilot study showed utility of plasmapheresis and bortezomib in reduction of cPRA.63 Use of plasmapheresis and bortezomib in this context followed the protocol used for treatment of acute rejection post-kidney transplant. Infectious risk may be higher in patients who undergo desensitization treatment. It is uncertain if the benefit of therapy outweighs the risk of infection due to desensitization strategies.
Immediately post-heart transplant, induction therapy can be used for sensitized patients to potentially reduce their risk for rejection. A number of different modalities have been used in an induction strategy. Common induction agents include T-cell-depleting agents, including ATG, and IL-2 receptor antagonists, including basiliximab (Simulect®; Novartis International AG). Routine induction therapy due to physician choice occurs in approximately 50% of heart transplants. The use of routine induction therapy at the time of heart transplant is controversial, as there is no clear survival benefit with its use.
Another strategy under current investigation, which may serve as an alternative to induction therapy, involves use of the terminal complement inhibitor eculizumab (Soliris®; Alexion Pharmaceuticals, Cheshire, CT, USA) at the time of heart transplant and protocol-based administration in the first 2 months post-transplant. Eculizumab is a humanized anti-C5 antibody which impairs C5 cleavage to C5a and C5b. This inhibition prevents formation of the membrane attack complex (MAC) and C5a-induced chemotaxis of inflammatory cells. Cd4 deposition, C3a activity, and other upstream complement cascade steps are not affected. Eculizumab significantly reduced, but did not abolish, early AMR in renal transplant recipients with known DSA against their living related donor.64 This suggests the possibility of complement-independent mechanisms of acute AMR. There did not appear to be significant infectious risk with use of this agent. Since the complement cascade acts as an intermediary between the innate and the adaptive immune systems, there is a chance that terminal complement inhibition will modulate the immune system of patients post-transplant, reduce rates of AMR, and possibly impact CAV.
Further understanding of the mechanisms of AMR and CAV will potentially lead to therapeutic targets to prevent the development or slow the process of CAV. Antigens of various endothelial cells, including angiotensin II type 1 receptor,65 anti-MHC class I chain-related A (MICA),66 anti-MHC class I chain-related B (MICB), vimentin, and adhesion or trafficking receptors may play a role in CAV development. Inflammatory modulators that affect cytokine signaling may have a mechanistic role in the development of CAV.67,68
Conclusion
Cardiac allograft immune activation and heart transplant rejection remain major contributors toward morbidity and mortality after heart transplant. Antibody sensitization presents additional challenges in the care of heart transplant recipients. Further understanding of the interactions between the innate and adaptive immune systems is key to effective immunomodulation and prevention of cardiac allograft activation and heart transplant rejection. Targeted therapies could lead to improved patient care with improved graft and patient outcomes and reduced treatment side effects. Complement deposition is part of the process of AMR. The utility of terminal complement inhibition is under current examination and represents one example of modulation of the innate and adaptive immune systems.
Disclosure
Dr Kobashigawa has received honoraria and/or research grants from Novartis Pharmaceuticals and CareDx Inc. Dr Chang owns stock in the company AbbVie. The authors report no other conflicts of interest in this work.
References
Adler ED, Goldfinger JZ, Kalman J, Park ME, Meier DE. Palliative care in the treatment of advanced heart failure. Circulation. 2009; 120:2597–2606. | |
Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med. 2001;345:1435–1443. | |
Stehlik J, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Twenty-eighth Adult Heart Transplant Report – 2011. J. Heart Lung Transplant. 2011;30:1078–1094. | |
Reed EF, Demetris AJ, Hammond E, et al; International Society for Heart and Lung Transplantation. Acute antibody-mediated rejection of cardiac transplants. J Heart Lung Transplant. 2006;25(2):153–159. | |
Benichou G, Tonsho M, Tocco G, Nadazdin O, Madsen JC. Innate immunity and resistance to tolerogenesis in allotransplantation. Front Immunol. 2012;3:73. | |
Vallejo JG. Role of toll-like receptors in cardiovascular diseases. Clin Sci (Lond). 2011;121(1):1–10. | |
Asea A. Heat shock proteins and toll-like receptors. In: Bauer S, Hartmann G, editors. Toll-Like Receptors (TLRs) and Innate Immunity. Berlin: Springer; 2008:111–127. | |
Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300(5625):1524–1525. | |
Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–2859. | |
Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425–435. | |
Vieyra M, Leisman S, Raedler H, et al. Complement regulates CD4 T-cell help to CD8 T cells required for murine allograft rejection. Am J Pathol. 2011;179(2):766–774. | |
Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 2013;34(2):74–80. | |
Issa F, Robb RJ, Wood KJ. The where and when of T cell regulation in transplantation. Trends Immunol. 2013;34(3):107–113. | |
Murphy SP, Porrett PM. Turka LA. Innate immunity in transplant tolerance and rejection. Immunol Rev. 2011;241(1):39–48. | |
Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2011;9(3):160–167. | |
Pikul FJ, Bolman RM, Saffitz JE, Chaplin H. Anti-B-mediated rejection of an ABO-incompatible cardiac allograft despite aggressive plasma exchange transfusion. Transplant Proc. 1987;19(6):4601–4604. | |
Patel R, Terasaki PI. Significance of the positive crossmatch test in kidney transplantation. N Engl J Med. 1969;280(14):735–739. | |
Terasaki PI. A personal perspective: 100-year history of the humoral theory of transplantation. Transplantation. 2012;93(8):751–756. | |
Bray RA, Nolen JD, Larsen C, et al. Transplanting the highly sensitized patient: the emory algorithm. Am J Transplant. 2006;6(10):2307–2315. | |
Stehlik J, Islam N, Hurst D, et al. Utility of virtual crossmatch in sensitized patients awaiting heart transplantation. J Heart Lung Transplant. 2009;28(11):1129–1134. | |
Pei R, Lee JH, Shih NJ, Chen M, Terasaki PI. Single human leukocyte antigen flow cytometry beads for accurate identification of human leukocyte antigen antibody specificities. Transplantation. 2003;75(1):43–49. | |
Cecka JM. Calculated PRA (CPRA):the new measure of sensitization for transplant candidates. Am J Transplant. 2010;10(1):26–29. | |
Kobashigawa J, Mehra M, West L, et al; Consensus Conference Participants. Report from a consensus conference on the sensitized patient awaiting heart transplantation. J Heart Lung Transplant. 2009;28(3):213–225. | |
Sutherland SM, Chen G, Sequeira FA, Lou CD, Alexander SR, Tyan DB. Complement-fixing donor-specific antibodies identified by a novel C1q assay are associated with allograft loss. Pediatr Transplant. 2012;16(1):12–17. | |
Caves P, Coltart J, Billingham M, Rider A, Stinson E. Transvenous endomyocardial biopsy – application of a method for diagnosing heart disease. Postgrad Med J. 1975;51(595):286–290. | |
Stewart S, Winters GL, Fishbein MC, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant. 2005;24(11):1710–1720. | |
Jarcho J, Naftel DC, Shroyer TW, et al. Influence of HLA mismatch on rejection after heart transplantation: a multiinstitutional study. The Cardiac Transplant Research Database Group. J Heart Lung Transplant. 1994;13(4):583–595. | |
Herskowitz A, Soule LM, Ueda K, et al. Arteriolar vasculitis on endomyocardial biopsy: a histologic predictor of poor outcome in cyclosporine-treated heart transplant recipients. J Heart Transplant. 1987;6(3):127–136. | |
Kobashigawa J, Crespo- Leiro MG, Ensminger SM, et al; Consensus Conference Participants. Report from a consensus conference on antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. 2011;30(3):252–269. | |
Berry GJ, Angelini A, Burke MM, et al. The ISHLT working formulation for pathologic diagnosis of antibody-mediated rejection in heart transplantation: evolution and current status (2005–2011). J Heart Lung Transplant. 2011;30(6):601–611. | |
Smith JD, Banner NR, Hamour IM, et al. De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant. 2011;11(2):312–319. | |
Tambur AR, Pamboukian SV, Costanzo MR, et al. The presence of HLA-directed antibodies after heart transplantation is associated with poor allograft outcome. Transplantation. 2005;80(8):1019–1025. | |
Taylor DO, Yowell RL, Kfoury AG, Hammond EH, Renlund DG. Allograft coronary artery disease: clinical correlations with circulating anti-HLA antibodies and the immunohistopathologic pattern of vascular rejection. J Heart Lung Transplant. 2000;19(6):518–521. | |
Patel JK, Kobashigawa JA. Improving survival during heart transplantation: diagnosis of antibody-mediated rejection and techniques for the prevention of graft injury. Future Cardiol. 2012;8(4):623–635. | |
Wu GW, Kobashigawa JA, Fishbein MC, et al. Asymptomatic antibody-mediated rejection after heart transplantation predicts poor outcomes. J Heart Lung Transplant. 2009;28(5):417–422. | |
Mehra MR, Crespo-Leiro MG, Dipchand A, et al. Erratum. J Heart Lung Transplant. 2011;30(3):360. | |
Kobashigawa JA, Tobis JM, Starling RC, et al. Multicenter intravascular ultrasound validation study among heart transplant recipients: outcomes after five years. J Am Coll Cardiol. 2005;45(9):1532–1537. | |
Fang JC, Kinlay S, Beltrame J, et al. Effect of vitamins C and E on progression of transplant-associated arteriosclerosis: a randomised trial. Lancet. 2002;359(9312):1108–1113. | |
Romeo G, Houyel L, Angel CY, Brenot P, Riou JY, Paul J. Coronary stenosis detection by 16-slice computed tomography in heart transplant patients: comparison with conventional angiography and impact on clinical management. J Am Coll Cardiol. 2005;45(11):1826–1831. | |
Wever-Pinzon O, Romero J, Kelesidis I, et al. Coronary computed tomography angiography for the detection of cardiac allograft vasculopathy: a meta-analysis of prospective trials. J Am Coll Cardiol. 2014;63(19):1992–2004. | |
Kobashigawa J. Coronary computed tomography angiography: is it time to replace the conventional coronary angiogram in heart transplant patients? J Am Coll Cardiol. 2014;63(19):2005–2006. | |
Wu YW, Chen YH, Wang SS, et al. PET assessment of myocardial perfusion reserve inversely correlates with intravascular ultrasound findings in angiographically normal cardiac transplant recipients. J Nucl Med. 2010;51(6):906–912. | |
Hou J, Lv H, Jia H, et al. OCT assessment of allograft vasculopathy in heart transplant recipients. JACC Cardiovasc Imaging. 2012;5(6):662–663. | |
Wu YL, Ye Q, Ho C. Cellular and functional imaging of cardiac transplant rejection. Curr Cardiovasc Imaging Rep. 2011;4(1):50–62. | |
Bader FM, Islam N, Mehta NA, et al. Noninvasive diagnosis of cardiac allograft rejection using echocardiography indices of systolic and diastolic function. Transplant Proc. 2011;43(10):3877–3881. | |
Pham MX, Teuteberg JJ, Kfoury AG, et al; IMAGE Study Group. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362:1890–1900. | |
Pham MX, Deng MC, Kfoury AG, Teuteberg JJ, Starling RC, Valantine H. Molecular testing for long-term rejection surveillance in heart transplant recipients: design of the Invasive Monitoring Attenuation Through Gene Expression (IMAGE) trial. J Heart Lung Transplant. 2007;26(8):808–814. | |
Kobashigawa J, Patel J, Kittleson M, et al. Results of a randomized trial of Allomap vs heart biopsy in the 1st year after heart transplant: early invasive monitoring attenuation through gene expression trial. J Heart Lung Transplant. 2013;32(4):S203. | |
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation Guidelines for the care of heart transplant recipients. J Heart Lung Transplant. 2010;29(8):914–956. | |
Kobashigawa JA, Kiyosaki KK, Patel JK, et al. Benefit of immune monitoring in heart transplant patients using ATP production in activated lymphocytes. J Heart Lung Transplant. 2010;29(5):504–508. | |
Cheng R, Azarbal B, Patel J, Chang D, Kobashigawa J. Elevated immune monitoring after cardiac transplantation is associated with increased plaque progression by intravascular ultrasound. J Am Coll Cardiol. 2014;63(12_S). | |
Baran DA, Zucker MJ, Arroyo LH, et al. A prospective, randomized trial of single-drug versus dual-drug immunosuppression in heart transplantation: the tacrolimus in combination, tacrolimus alone compared (TICTAC) trial. Circ Heart Fail. 2011;4(2):129–137. | |
Kobashigawa JA. Strategies in immunosuppression after heart transplantation: is less better? Circ Heart Fail. 2011;4:111–113. | |
Kobashigawa JA, Miller LW, Russell SD, et al; Study Investigators. Tacrolimus with mycophenolate mofetil (MMF) or sirolimus vs cyclosporine with MMF in cardiac transplant patients: 1-year report. Am J Transplant. 2006;6(6):1377–1386. | |
Andreassen AK, Gude E, Sigurdardottir V, et al. Scandinavian Heart Transplant Everolimus De Novo Study with Early Calcineurin Inhibitor Avoidance (SCHEDULE). J Heart Lung Transplant. 2013;32(4):S134. | |
Eisen HJ, Tuzcu EM, Dorent R, et al; RAD B253 Study Group. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N Engl J Med. 2003;349(9):847–858. | |
Eisen HJ, Kobashigawa J, Starling RC, et al. Everolimus versus mycophenolate mofetil in heart transplantation: a randomized, multicenter trial. Am J Transplant. 2013;13(5):1203–1216. | |
Marfo K, Lu A, Ling M, Akalin E. Desensitization protocols and their outcome. Clin J Am Soc Nephrol. 2011;6(4):922–936. | |
Aranda JM Jr, Scornik JC, Normann SJ, et al. Acti-CD20 monoclonal antibody (rituximab) therapy for acute cardiac humoral rejection: a case report. Transplantation. 2002;73(6):907–910. | |
Everly MJ, Everly JJ, Susskind B, et al. Bortezomib provides effective therapy for antibody- and cell-mediated acute rejection. Transplantation. 2008;86(12):1754–1761. | |
Kittleson MM, Patel JK, Moriguchi JD, et al. Heart transplant recipients supported with extracorporeal membrane oxygenation: outcomes from a single-center experience. J Heart Lung Transplant. 2011;30(11):1250–1256. | |
Vo AA, Lukovsky M, Toyoda M, et al. Rituximab and intravenous immune globulin for desensitization during renal transplantation. N Engl J Med. 2008;359:242–251. | |
Patel J, Everly M, Chang D, Kittleson M, Reed E, Kobashigawa J. Reduction of alloantibodies via proteasome inhibition in cardiac transplantation. J Heart Lung Transplant. 2011;30(12):1320–1326. | |
Stegall MD, Diwan T, Raghavaiah S, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11(11):2405–2413. | |
Yousufuddin M, Haji S, Starling RC, et al. Cardiac angiotensin II receptors as predictors of transplant coronary artery disease following heart transplantation. Eur Heart J. 2004;25(5):377–385. | |
Kauke T, Kaczmarek I, Dick A, et al. Anti-MICA antibodies are related to adverse outcome in heart transplant recipients. J Heart Lung Transplant. 2009;28(4):305–311. | |
Qin L, Huang Q, Zhang H, et al. SOCS1 prevents graft arteriosclerosis by preserving endothelial cell function. J Am Coll Cardiol. 2014;63(1):21–29. | |
de Weger RA. Immune regulators regulated to prevent transplant reactions. J Am Coll Cardiol. 2014;63(1):30–32. | |
Chang DH, Kittleson MM, Kobashigawa JA. Immunosuppression following heart transplantation: prospects and challenges. Immunotherapy. 2014;6(2):181–194. | |
Berry GC, Burke MM, Andersen C et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. 2013;32(12):1147–1162. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.