")
Back to Journals » Vascular Health and Risk Management » Volume 11
Breast cancer therapy and cardiovascular risk: focus on trastuzumab
Authors Sandoo A, Kitas G, Carmichael A
Received 6 February 2015
Accepted for publication 3 March 2015
Published 7 April 2015 Volume 2015:11 Pages 223—228
DOI https://doi.org/10.2147/VHRM.S69641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Video abstract presented by Aamer Sandoo.
Views: 2308
Aamer Sandoo,1,2 George D Kitas,2 Amtul R Carmichael2
1School of Sport, Health and Exercise Sciences, Bangor University, Bangor, Wales, 2Dudley Group of Hospitals NHS Trust, Russells Hall Hospital, Dudley, UK
Abstract: Breast cancer is the most common cancer in the UK. Advances in the methods of early diagnosis as well as newer and more effective treatments have led to improvements of disease-free and overall survival over the last decade. Almost one-third of breast cancers present with an aggressive form characterized by increased expression of human epidermal growth receptor 2 (HER2) proteins. A targeted treatment using monoclonal antibodies against HER2 expression such as trastuzumab has been shown to improve survival. Unfortunately, there is a degree of cardiotoxicity associated with these agents, as inhibition of HER2 pathways can also impair cardioprotective pathways. In the present review, we discuss the mechanisms by which trastuzumab might affect vascular homeostasis leading to endothelial dysfunction. We also provide suggestions for future research examining the effects of trastuzumab on the vasculature in breast cancer.
Keywords: endothelial dysfunction, microvasculature, macrovasculature, congestive heart failure
Introduction
Breast cancer is the most common cancer in the UK and accounts for 30% of all new cancer cases in females with a greater incidence in women over the age of 60 years.1 Fortunately, a variety of treatments including surgical resection, adjuvant chemotherapy, hormonal therapy, and radiation therapy have been shown to improve survival and reduce the risk of tumor reoccurrence. Approximately 15%–25% of patients present with aggressive breast cancer characterized by increased expression of human epidermal growth receptor 2 (HER2) proteins in the breast tissue.1 It is postulated that HER2-positivity increases the likelihood of invasion and survival of tumor cells at the site of metastasis.2 Therefore, patients with HER2-positive tumors may also have increased resistance to common anticancer treatments, such as chemotherapy and radiation therapy.2
In these patients, targeted treatment using monoclonal antibodies such as trastuzumab (Herceptin) can be used to reduce tumor recurrence and improve survival.3,4 Trastuzumab selectively binds to the extracellular domain of HER2 receptors, where it inhibits downstream signaling pathways, resulting in a reduced proliferation of tumor cells. This is achieved by identifying tumor cells for immune destruction, and then, by initiation of antibody-dependent cellular cytotoxicity, causing apoptosis of tumor cells.5 In addition, trastuzumab undergoes internalization (endocytosis) into the tumor cells and subsequently increases the expression of HER2 on the cellular surface. This enhances the immune effects of trastuzumab and reduces tumor expression.5 However, trastuzumab is also associated with an increased risk of cardiotoxicity which manifests clinically as congestive heart failure (CHF).6 Trastuzumab-mediated cardiotoxicity appears to be independent of drug dose, has not been shown to be associated with structural changes in cardiomyocytes, and is fully reversible following cessation of treatment.7 Cardiotoxicity is more likely to occur in patients who have preexisting hypertension, a history of smoking, obesity, family history of cardiovascular disease (CVD), and previous coronary artery disease – all of which are well-established risk factors for cardiac events.8,9 CVD and its sequelae are strong predictors of mortality in patients with breast cancer, this association being independent of breast cancer stage.10 It is therefore possible that trastuzumab-related cardiotoxicity might be mediated by adverse drug effects on the coronary vasculature. In this review, we summarize the biological mechanisms by which trastuzumab may affect the vasculature and contribute to CVD risk. We also provide suggestions about the investigation of trastuzumab-mediated cardiotoxicity in future research studies.
The importance of HER2 and neuregulin
The human epidermal growth receptors are tyrosine-kinase receptors and are expressed as four isoforms: HER1, HER2, HER3, and HER4. The second isoform (HER2) regulates the growth, repair, and regeneration of breast tissue,11 but overexpression of HER2 receptors via polymorphisms in the erb-b2 receptor tyrosine kinase 2 oncogene can lead to uncontrolled cell growth.12 This state is known as HER2-positivity and is linked with increased mortality.13,14 Under normal conditions, a protein called neuregulin is released by coronary microvascular endothelial cells and the endocardium.15 Neuregulin binds to HER4 receptors which dimerize with HER2 receptors and increase several survival pathways in the myocardium.15 The primary role of the survival pathways is to inhibit the production of reactive oxygen species (ROS) and maintain cellular integrity by reducing cell apoptosis. In an animal model of adult rat ventricular myocytes, treatment with either paclitaxel or trastuzumab resulted in damage to cardiac myofilaments which corresponded with increased intracellular calcium, reduced diastolic relaxation time, and increased oxidative stress.16 These adverse changes following trastuzumab increase the risk of developing CHF (for review, see Sandoo et al17). Indeed, the HER2/neuregulin pathway is integral to the preservation of sarcomeres in the cardiomyocytes.18 However, neuregulin can also increase the expression of endothelial nitric oxide synthase (eNOS).15 The eNOS gene is constitutively expressed in vascular endothelial cells and releases nitric oxide (NO) – a vasoactive molecule that reduces oxidative stress and prevents atherosclerosis.19 NO bioavailability is increased via activation from protein kinase B as a result of the dimerization of HER2 and HER4 receptors.15 Trastuzumab blocks this dimerization and therefore inhibits the cardioprotective actions of neuregulin.20 Given that the myocardium is a highly vascularized territory, it is possible that abnormalities in the vasculature following trastuzumab could also contribute to CHF. In the following section, we discuss the implications of blocking HER2 using trastuzumab on vascular endothelial cells, and highlight how this may contribute to cardiovascular complications in breast cancer.
Mechanisms for trastuzumab-mediated endothelial dysfunction
eNOS
The endothelium is the delicate and permeable lining of the vasculature which allows specific vasoactive factors to regulate homeostatic processes and prevent atherosclerosis.19 In a healthy vessel, eNOS continuously synthesizes NO, helping to maintain basal vasodilator tone and preventing platelet aggregation and formation of ROS.19 In chronic disease states characterized by increased oxidative stress and low-grade chronic inflammation (such as breast cancer), endothelial cells can be damaged, leading to a reduction in eNOS expression and endothelial dysfunction.21,22
The expression of eNOS has been postulated to play an important role in regulating cardiovascular function and overall survival in breast cancer. In a recent study, Zeglinski et al23 compared 60 wild-type mice with 60 mice having a congenital absence of the eNOS gene treated with either saline, doxorubicin, or trastuzumab, or both drugs simultaneously. Following simultaneous treatment with both agents, the mice with absence of the eNOS gene presented with adverse modeling of the left ventricle, reduced left ventricular systolic function, increased oxidative stress, and reduced survival when compared with the wild-type mice. These findings suggest that adequate expression of the eNOS gene is important in suppressing the generation of ROS which may in turn improve cardiac function and survival following trastuzumab treatment. Indeed, in human breast cancer patients receiving chemotherapy, polymorphisms in the eNOS gene result in poorer survival,24 and expression of eNOS in the microvessels surrounding breast tumors associates with better prognosis, possibly due to a reduction in tumor progression.25 In addition, polymorphisms in the Asp298 allele of the eNOS gene result in impaired NO synthesis which reduces event-free survival in patients with systolic heart failure.26
Generation of ROS
Inhibition of the neuregulin/HER2 pathway by trastuzumab allows increased production of ROS,15 due to abnormalities in mitochondrial respiration in cardiomyocytes, which increases myocardial stress.27 When the myocardium is increasingly stressed, angiotensin II (ANG II), thrombin, platelet-derived growth factor, and tumor growth factor-alpha are upregulated and stimulate vascular smooth muscle cells to increase formation of further ROS.22 ANG II, which is a potent vasoconstrictor, also inhibits neuregulin activity in the coronary microvasculature and increases levels of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH).28,29 NADPH has been implicated as a causal factor of endothelial dysfunction due to its ability to generate large amounts of ROS in the endothelium.22 The mechanisms highlighted above represent a vicious cycle of free radical formation which will injure the endothelium causing endothelial dysfunction (Figure 1).
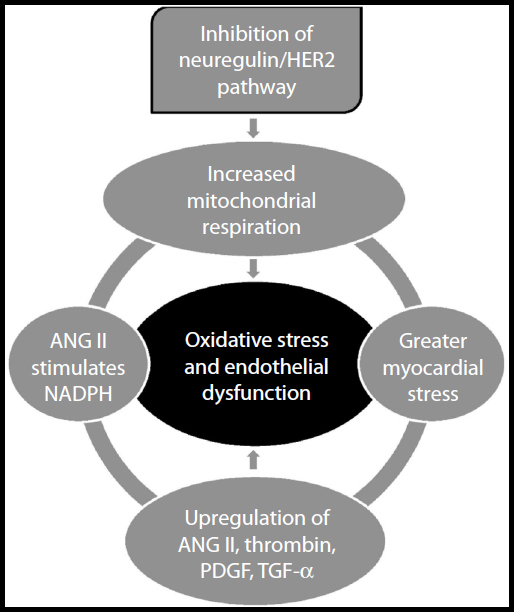 | Figure 1 The generation of reactive oxygen species following inhibition of the neuregulin/HER2 pathway. |
Endothelial dysfunction and CHF
Treatment with trastuzumab does not cause morphological alterations to cardiomyocytes, and myocardial function can improve following cessation of treatment.30 Although apoptosis of cardiomyocytes following trastuzumab treatment can result in CHF,31 in the general population, endothelial dysfunction is one of the primary causal factors for CHF.32–35 On this basis, it has been hypothesized that endothelial dysfunction might be the principal mechanism for trastuzumab- mediated CHF (see Figure 2).17
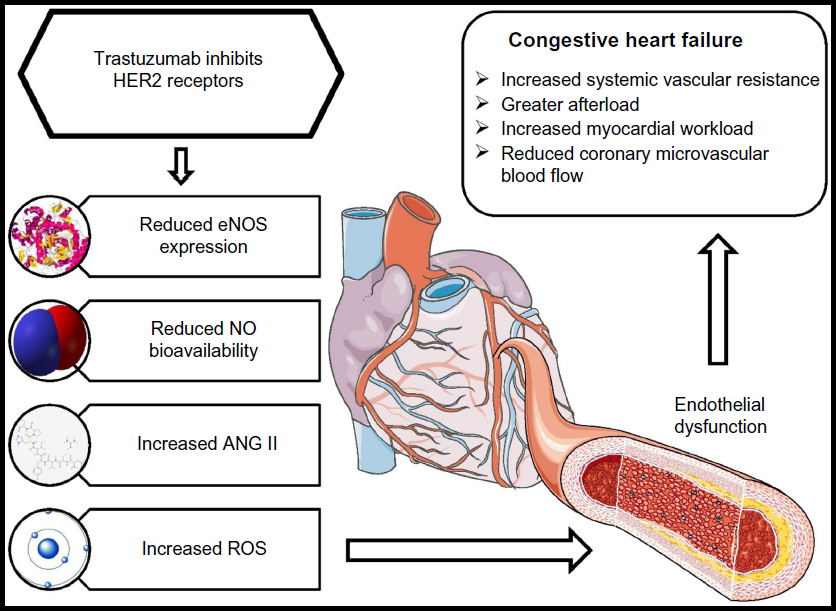 | Figure 2 The effects of trastuzumab on the vasculature and its subsequent contribution to congestive heart failure. |
The myocardial microvasculature receives less than 5% of cardiac output yet regulates the majority of blood flow to the myocardium.36 Unsurprisingly, in a 5-year prospective study of 242 CHF patients, microvascular endothelial dysfunction was independently associated with cardiac events.33 Similarly, Fischer et al32 revealed that large-vessel endothelial dysfunction was independently associated with higher incidence of hospitalization from CHF, cardiac transplantation, and cardiac death. These findings suggest that global alterations in the vasculature are important determinants for the development of CHF and associated comorbidities.
The central mechanisms in the development of CHF involve the reduction in L-arginine and tetrahydrobiopterin (BH4) concentrations – both essential substrates for the synthesis of NO.37 In particular, absence of the NO cofactor BH4 can increase the production of ROS, perpetuating myocardial damage. The conversion of L-arginine to NO by eNOS requires the presence of BH4 which helps the transfer of electrons produced at the eNOS site to an oxidase domain, resulting in activation of oxygen. The activated oxygen is then “coupled” with L-arginine oxidation to produce NO.38 However, in the absence of BH4 or L-arginine, eNOS produces superoxide molecules instead of NO.38 Levels of BH4 are reduced in CHF,39 and the consequent reduction in NO will impact on coronary vasorelaxation and basal blood flow, both of which are important physiological factors for the development of CHF.40
The renin–angiotensin system also plays an important role in regulating the release of NO from vascular endothelial cells. In CHF patients presenting with decreased vasodilatory function of the large arteries, angiotensin-converting enzyme (ACE) inhibitors improve vasodilatation as a result of increased NO bioavailability.41 Similarly, endothelial function improves following administration of ANG II receptor antagonists concomitantly with ACE inhibitors.42 In breast cancer patients treated with trastuzumab, concomitant administration of ACE inhibitors, ANG II receptor antagonists, and beta-blockers improves left ventricular ejection fraction over the course of treatment.43 Given that trastuzumab can reduce NO production and increase production of ANG II via inhibition of neuregulin, it is plausible that CHF resulting from trastuzumab treatment is likely to be mediated, in part, by endothelial dysfunction of the coronary vasculature.
Role of classical CVD risk in potentiating trastuzumab-mediated cardiotoxicity
The link between endothelial dysfunction and development of CHF in trastuzumab-treated patients could be mediated by several classical CVD risk factors. Associations between CVD risk factors implicated in endothelial dysfunction and trastuzumab-mediated cardiotoxicity have been reported.8,9 The presence of severe hypertension in particular appears to be an important factor in the development of heart failure following trastuzumab treatment.8,44 Hypertension usually occurs when NO bioavailability is reduced, allowing vasoconstrictors such as endothelin-1 and ANG II to increase systemic vascular resistance.45 The direct consequence is an augmentation in afterload and intracavitary pressures of the left ventricle. If the increased myocardial stress is coupled with poor myocardial blood flow, cardiac workload will be increased, leading to profound damage of the myocardium and CHF.46 Thus, the impact of CVD risk status is important in understanding the impact of trastuzumab treatment on the vasculature.
Proposals for future research studies
The investigation of endothelial dysfunction following trastuzumab treatment has received little attention in the literature to date, yet it is clear that abnormalities in the vasculature following inhibition of HER2 could be implicated in the development of trastuzumab-mediated cardiotoxicity. At present, several noninvasive assessments which examine vasodilatory function and arterial structure can be utilized in patients with breast cancer.19 These peripheral vascular assessments correlate well with assessments in the coronary circulation47,48 and are strong predictors of cardiac events in a variety of clinical populations at risk of CVD,49,50 including postmenopausal women.51 Thus, we suggest that, in addition to further in vitro and animal model work, studies should also be conducted to 1) identify whether there is evidence of endothelial dysfunction in trastuzumab-treated patients when compared to non-trastuzumab-treated patients and healthy controls; 2) establish the time course of endothelial dysfunction by conducting long-term prospective studies examining endothelial function at pretreatment baseline and at multiple follow-up points; and 3) assess the impact of classical CVD risk factors and their control on the above. Such studies will enable better understanding of the processes that cause CHF following trastuzumab treatment, inform possible pharmacological and nonpharmacological interventions, and eventually improve cardiovascular outcomes in this patient population.
Disclosure
The authors report no conflicts of interest in this work.
References
Breast cancer statistics [webpage on the internet]. London: Cancer Research UK; [updated November 27, 2014). Available from: http://www.cancerresearchuk.org/cancer-info/cancerstats/types/breast/. Accessed January 15, 2015. | |
Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene. 2000;19:6115–6121. | |
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. | |
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al; Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. | |
Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61:4744–4749. | |
Zeglinski M, Ludke A, Jassal DS, Singal PK. Trastuzumab-induced cardiac dysfunction: a ‘dual-hit’. Exp Clin Cardiol. 2011;16:70–74. | |
Floyd JD, Nguyen DT, Lobins RL, Bashir Q, Doll DC, Perry MC. Cardiotoxicity of cancer therapy. J Clin Oncol. 2005;23:7685–7696. | |
Wadhwa D, Fallah-Rad N, Grenier D, et al. Trastuzumab mediated cardiotoxicity in the setting of adjuvant chemotherapy for breast cancer: a retrospective study. Breast Cancer Res Treat. 2009;117:357–364. | |
Gunaldi M, Duman BB, Afsar CU, et al. Risk factors for developing cardiotoxicity of trastuzumab in breast cancer patients: an observational single-centre study. J Oncol Pharm Pract. Epub January 7, 2015. | |
Satariano WA, Ragland DR. The effect of comorbidity on 3-year survival of women with primary breast cancer. Ann Intern Med. 1994;120:104–110. | |
Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. | |
Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;11:1936–1942. | |
Berchuck A, Kamel A, Whitaker R, et al. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Res. 1990;50:4087–4091. | |
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. | |
Lemmens K, Doggen K, De Keulenaer GW. Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease: implications for therapy of heart failure. Circulation. 2007;116:954–960. | |
Pentassuglia L, Timolati F, Seifriz F, Abudukadier K, Suter TM, Zuppinger C. Inhibition of ErbB2/neuregulin signaling augments paclitaxel-induced cardiotoxicity in adult ventricular myocytes. Exp Cell Res. 2007;313:1588–1601. | |
Sandoo A, Kitas GD, Carmichael AR. Endothelial dysfunction as a determinant of trastuzumab-mediated cardiotoxicity in patients with breast cancer. Anticancer Res. 2014;34:1147–1151. | |
Kuramochi Y, Guo X, Sawyer DB. Neuregulin activates erbB2-dependent src/FAK signaling and cytoskeletal remodeling in isolated adult rat cardiac myocytes. J Mol Cell Cardiol. 2006;41:228–235. | |
Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302–312. | |
Pentassuglia L, Sawyer DB. The role of Neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res. 2009;315:627–637. | |
Wilcox JN, Subramanian RR, Sundell CL, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17:2479–2488. | |
Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. | |
Zeglinski M, Premecz S, Lerner J, et al. Congenital absence of nitric oxide synthase 3 potentiates cardiac dysfunction and reduces survival in doxorubicin- and trastuzumab-mediated cardiomyopathy. Can J Cardiol. 2014;30:359–367. | |
Choi JY, Barlow WE, Albain KS, et al. Nitric oxide synthase variants and disease-free survival among treated and untreated breast cancer patients in a Southwest Oncology Group clinical trial. Clin Cancer Res. 2009;15:5258–5266. | |
Mortensen K, Holck S, Christensen IJ, et al. Endothelial cell nitric oxide synthase in peritumoral microvessels is a favorable prognostic indicator in premenopausal breast cancer patients. Clin Cancer Res. 1999;5:1093–1097. | |
McNamara DM, Holubkov R, Postava L, et al. Effect of the Asp298 variant of endothelial nitric oxide synthase on survival for patients with congestive heart failure. Circulation. 2003;107:1598–1602. | |
Grazette LP, Boecker W, Matsui T, et al. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J Am Coll Cardiol. 2004;44:2231–2238. | |
Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851–859. | |
Cardinale D, Colombo A, Sandri MT, et al. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation. 2006;114:2474–2481. | |
Di Cosimo S. Heart to heart with trastuzumab: a review on cardiac toxicity. Target Oncol. 2011;6:189–195. | |
Gordon LI, Burke MA, Singh AT, et al. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J Biol Chem. 2009;284:2080–2087. | |
Fischer D, Rossa S, Landmesser U, et al. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. 2005;26:65–69. | |
de Berrazueta JR, Guerra-Ruiz A, García-Unzueta MT, et al. Endothelial dysfunction, measured by reactive hyperaemia using strain-gauge plethysmography, is an independent predictor of adverse outcome in heart failure. Eur J Heart Fail. 2010;12:477–483. | |
Hornig B, Maier V, Drexler H. Physical training improves endothelial function in patients with chronic heart failure. Circulation. 1996;93:210–214. | |
Hornig B, Arakawa N, Kohler C, Drexler H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation. 1998;97:363–368. | |
Brutsaert DL, Fransen P, Andries LJ, De Keulenaer GW, Sys SU. Cardiac endothelium and myocardial function. Cardiovasc Res. 1998;38:281–290. | |
Linke A, Recchia F, Zhang X, Hintze TH. Acute and chronic endothelial dysfunction: implications for the development of heart failure. Heart Fail Rev. 2003;8:87–97. | |
Alkaitis MS, Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep. 2012;9:200–210. | |
Moens AL, Takimoto E, Tocchetti CG, et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. | |
Seddon M, Melikian N, Dworakowski R, et al. Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation. 2009;119:2656–2662. | |
Hornig B, Arakawa N, Haussmann D, Drexler H. Differential effects of quinaprilat and enalaprilat on endothelial function of conduit arteries in patients with chronic heart failure. Circulation. 1998;98:2842–2848. | |
Giannattasio C, Achilli F, Grappiolo A, et al. Radial artery flow-mediated dilatation in heart failure patients: effects of pharmacological and nonpharmacological treatment. Hypertension. 2001;38:1451–1455. | |
Oliva S, Cioffi G, Frattini S, et al; Italian Cardio-Oncological Network. Administration of angiotensin-converting enzyme inhibitors and β-blockers during adjuvant trastuzumab chemotherapy for nonmetastatic breast cancer: marker of risk or cardioprotection in the real world? Oncologist. 2012;17:917–924. | |
Herrmann J, Herrmann SM, Haddad TC. New-onset heart failure in association with severe hypertension during trastuzumab therapy. Mayo Clin Proc. 2014;89:1734–1739. | |
Nadar S, Blann AD, Lip GY. Endothelial dysfunction: methods of assessment and application to hypertension. Curr Pharm Des. 2004;10:3591–3605. | |
Buus NH, Bøttcher M, Hermansen F, Sander M, Nielsen TT, Mulvany MJ. Influence of nitric oxide synthase and adrenergic inhibition on adenosine-induced myocardial hyperemia. Circulation. 2001;104:2305–2310. | |
Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. | |
Takase B, Hamabe A, Satomura K, et al. Close relationship between the vasodilator response to acetylcholine in the brachial and coronary artery in suspected coronary artery disease. Int J Cardiol. 2005;105:58–66. | |
Gokce N, Keaney JF Jr, Hunter LM, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol. 2003;41:1769–1775. | |
Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. | |
Rossi R, Nuzzo A, Origliani G, Modena MG. Prognostic role of flow-mediated dilation and cardiac risk factors in post-menopausal women. J Am Coll Cardiol. 2008;51:997–1002. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.