")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties
Authors Wang Y, Yang Y, Zhe H, He Z, Zhou S
Received 5 June 2014
Accepted for publication 15 July 2014
Published 23 October 2014 Volume 2014:8 Pages 2075—2088
DOI https://doi.org/10.2147/DDDT.S68872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yan-Yang Wang,1,2 Yin-Xue Yang,3 Hong Zhe,1 Zhi-Xu He,4 Shu-Feng Zhou2,4
1Department of Radiation Oncology, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Department of Colon-rectal Surgery, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 4Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, Guizhou, People’s Republic of China
Abstract: Triterpenoids have been used for medicinal purposes in many Asian countries because of their anti-inflammatory, antioxidant, antiproliferative, anticancer, and anticarcinogenic properties. Bardoxolone methyl, the C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) known as CDDO-Me or RTA 402, is one of the derivatives of synthetic triterpenoids. CDDO-Me has been used for the treatment of chronic kidney disease, cancer (including leukemia and solid tumors), and other diseases. In this review, we will update our knowledge of the clinical pharmacokinetics and pharmacodynamics of CDDO-Me, highlighting its clinical benefits and the underlying mechanisms involved. The role of the Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1)/the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway in the therapeutic activities of CDDO-Me will be discussed. CDDO-Me contains a,ß-unsaturated carbonyl groups on rings A and C that can generate reversible adducts with the thiol groups of Cys residues in target proteins such as Keap1 and IκB kinase. At low nanomolar concentrations, CDDO-Me protects the cells against oxidative stress via inhibition of reactive oxygen species generation, while CDDO-Me at low micromolar concentrations induces apoptosis by increasing reactive oxygen species and decreasinging intracellular glutathione levels. Through Keap1/Nrf2 and nuclear factor-κB pathways, this agent can modulate the activities of a number of important proteins that regulate inflammation, redox balance, cell proliferation and programmed cell death. In a Phase I trial in cancer patients, CDDO-Me was found to have a slow and saturable oral absorption, a relatively long terminal phase half-life (39 hours at 900 mg/day), nonlinearity (dose-dependent) at high doses (600–1,300 mg/day), and high interpatient variability. As a multifunctional agent, CDDO-Me has improved the renal function in patients with chronic kidney disease associated with type 2 diabetes. CDDO-Me has shown a promising anticancer effect in a Phase I trial. This agent is generally well tolerated, but it may increase adverse cardiovascular events. Presently, it is being further tested for the treatment of patients with chronic kidney disease, cancer, and pulmonary arterial hypertension.
Keywords: CDDO-Me, chronic kidney disease, cancer, pulmonary arterial hypertension, Nrf2, Keap1, NF-κB
Introduction
Triterpenoids are a large family of compounds synthesized in some plants, such as the chrysanthemum flower through the cyclization of squalene, that have been used in traditional Asian medicine for disease management.1,2 Naturally occurring triterpenoids like oleanolic acid (OA) and ursolic acid have only weak anti-inflammatory and anticarcinogenic activities. To increase their usefulness, a series of novel derivatives of OA have been synthesized.3 The synthetic triterpenoids, such as 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) and its derivatives, such as CDDO methyl ester (CDDO-Me), the methyl amide of CDDO (CDDO-Ma), the ethyl amide of CDDO (CDDO-Ea), and CDDO-imidazolide (CDDO-Im), are compounds originally developed for the prevention and treatment of inflammation and cancer.4,5 Their chemical structures are shown in Figure 1. These synthetic triterpenoids are potent inhibitors of the de novo synthesis of inflammatory enzymes such as inducible nitric oxide synthase and inducible cyclooxygenase 2.4,6,7 Bardoxolone methyl, also known as CDDO-Me or RTA 402, is more potent than CDDO in anticancer and cancer-preventive activities8–11 and in the activation of Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1/nuclear factor erythroid 2-related factor 2/antioxidant response element (Keap1/Nrf2/ARE) pathway,12,13 which is involved in cytoprotection in the presence of excessive electrophiles or oxidative stress.
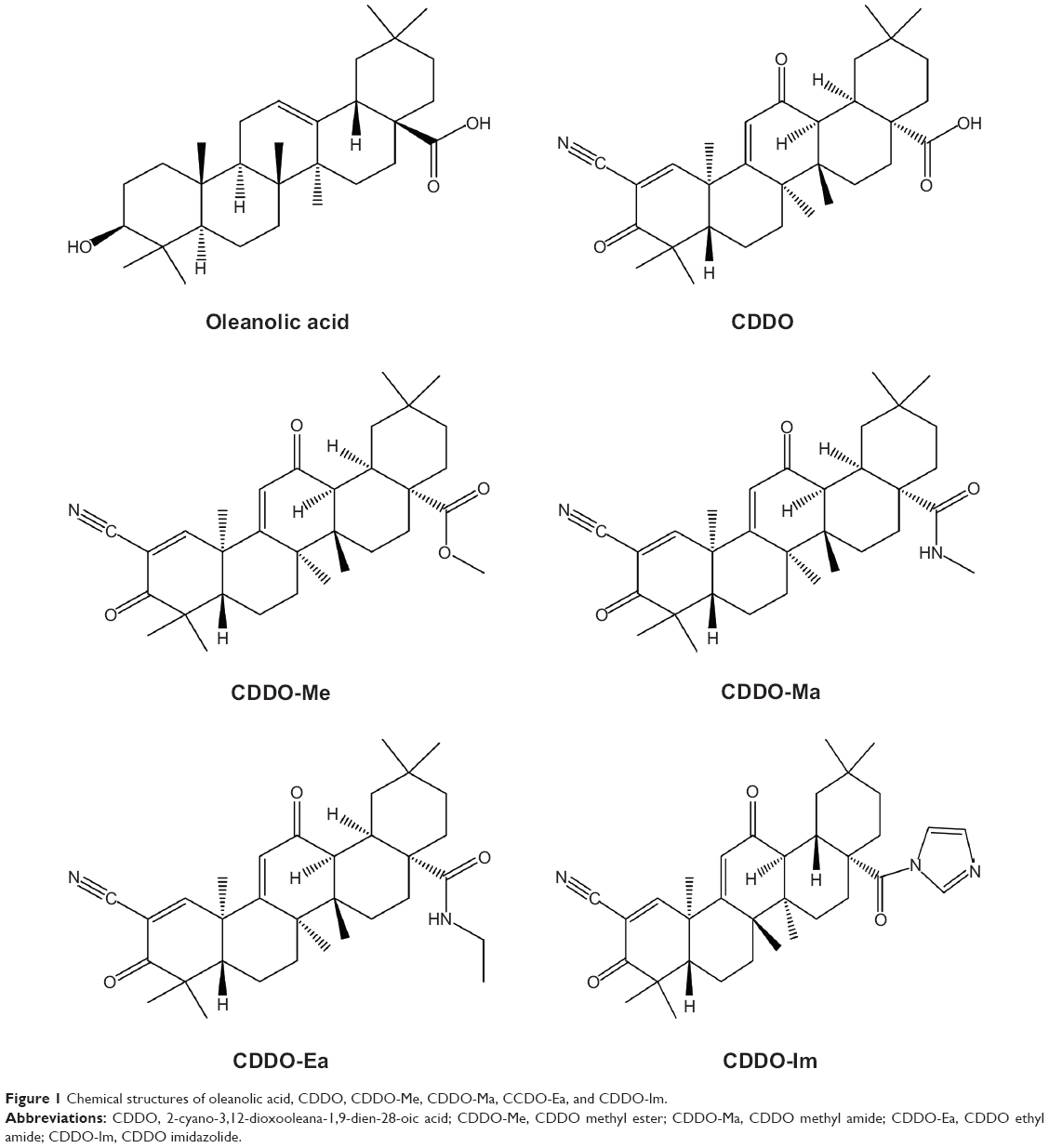 | Figure 1 Chemical structures of oleanolic acid, CDDO, CDDO-Me, CDDO-Ma, CCDO-Ea, and CDDO-Im. |
As a noncytotoxic and multifunctional drug, CDDO-Me has applications for the prevention and treatment of not only cancer, but also of many other diseases with an inflammatory component.14,15 The therapeutic effects of CDDO-Me have been tested in Phase III clinical trials for chronic kidney disease (CKD)16 and Phase I/II clinical trials for malignant diseases.17,18 In this review, we will update our knowledge of the pharmacokinetics, therapeutic effects, and mechanisms of action of CDDO-Me when used for the treatment of CKD, cancer, and other diseases. Both the clinical evidence and underlying mechanisms will be highlighted.
CDDO-Me behaves as a potent Nrf2 activator and nuclear factor-κB (NF-κB) inhibitor
Chemical modifications of OA on its three sites, the C-3 hydroxyl group, the C-12–C-13 double bond, and the C-28 carboxylic acid, have given rise to a series of new synthetic oleanane triterpenoids in order to increase their bioactivities.19,20 CDDO, CDDO-Me, and CDDO-Im possess better anti-inflammatory and cytoprotective activities than OA.14,15
The molecular mechanism of action of the triterpenoids is believed to be mediated by the Michael addition with active nucleophilic groups on proteins, such as the thiol groups on cysteine residues. In the development of OA derivatives, two electrophilic Michael acceptor sites were incorporated in the A and C rings of OA. Structure-activity analyses have shown that α,β-unsaturated carbonyl groups at key positions on rings A and C are essential for maintaining the potent anti-inflammatory activity of synthetic triterpenoids.13,21 In cells, synthetic oleanane triterpenoids mediate their potent pharmacological effects in part through interactions with cellular nucleophiles such as discrete, redox-sensitive sulfhydryl groups of cysteine residues on proteins via reversible Michael addition.19,22 Therefore, one may expect that synthetic oleanane triterpenoids should have many cellular protein binding partners, depending on the cellular context and the nucleophilicity of specific cysteine residues in individual targets.
Initial screening studies for determining specific targets for CDDO and related derivatives have shown that both CDDO and CDDO-Me bind to peroxisome-activated receptor γ and exhibit partial agonist and antagonist activities.4,23 However, many of the actions of synthetic oleanane triterpenoids occur independently of this notoriously promiscuous nuclear receptor.14 Other direct molecular targets of CDDO-Me include Keap1 and IκB kinase (IKKβ). The structure of CDDO-Me is similar to 15-deoxy-Δ12,14-prostaglandin J2 and related cyclopentenone prostaglandins, which are the endogenous activators of Nrf2 that play an important role in the resolution of inflammation and suppresses the activity of NF-κB (Figure 2).24–27 Binding of CDDO-Me to Keap1 disrupts its critical cysteine residues, leading to the release of Nrf2, which hinders its ubiquitination and finally leads to stabilization and nuclear translocation of NF-κB. In the nucleus, Nrf2 activates the transcription of phase 2 response genes, leading to a coordinated antioxidant and anti-inflammatory response.13 CDDO-Me also can bind to Cys-179 in the IKKβ activation loop and inhibit NF-κB activation.28 Binding of CDDO-Me to IKKβ prevents the release of NF-κB from its bound complex with IκB in the cytosol, thereby inhibiting NF-κB activation and downstream proinflammatory signaling pathways.28 It is important to recognize that binding of CDDO-Me to cysteine residues in specific proteins is both context-dependent and dose-dependent, which will be discussed later in this review. The nucleophilicity of the free sulfhydryl (-SH) groups in cysteine in a protein is markedly influenced by neighboring amino acids. Moreover, the unique stereochemistry of the oleanane scaffold prevents CDDO-Me from randomly alkylating protein targets.19
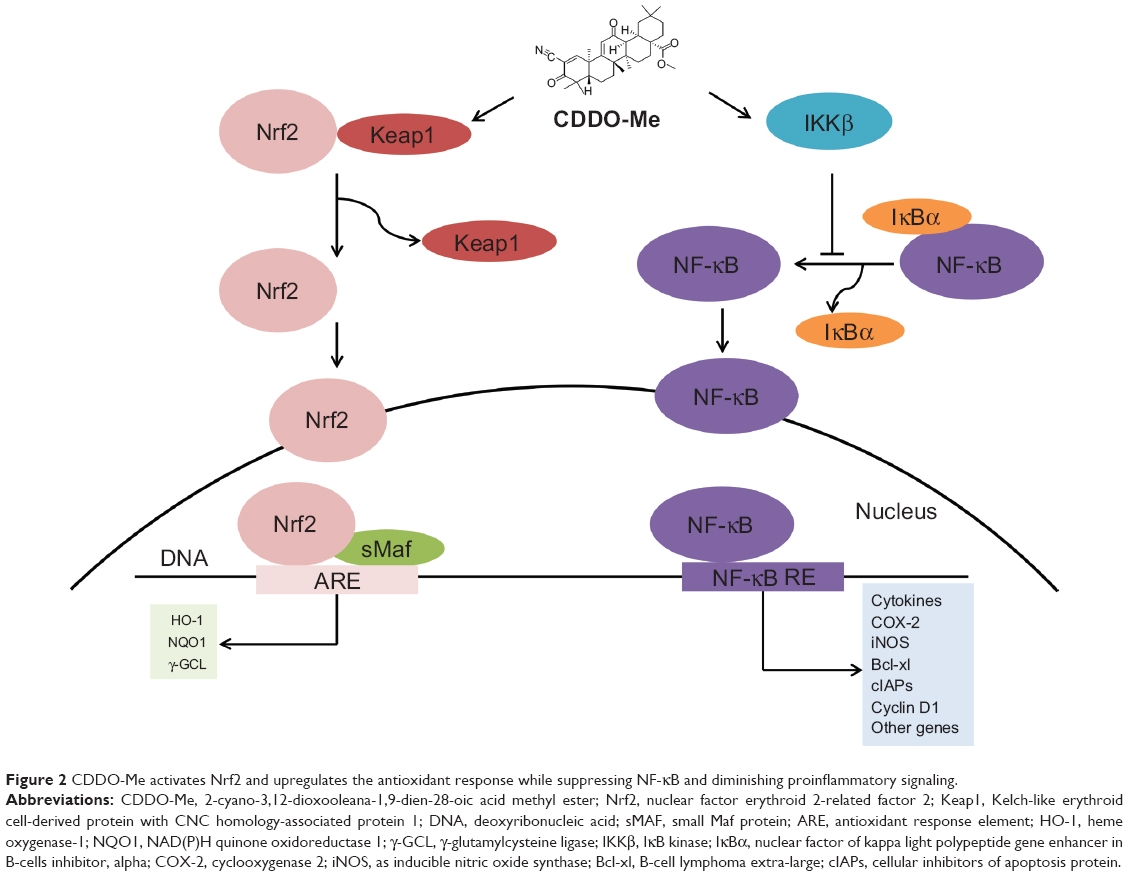 | Figure 2 CDDO-Me activates Nrf2 and upregulates the antioxidant response while suppressing NF-κB and diminishing proinflammatory signaling. |
A recent proteomic study in wild-type and Nrf2−/− mice has also provided evidence that CDDO-Me selectively activates the Keap1/Nrf2 pathway.29 Treatment of the wild-type mice with CDDO-Me at 3 mg/kg by intraperitoneal (ip) injection resulted in significantly altered expression of 43 proteins. Six proteins were regulated at both basal and inducible levels: cytochrome P450 2A5 (17.2-fold), glutathione S-transferase-Mu 3 (6.4-fold), glutathione S-transferase Mu 1 (5.9-fold), ectonucleoside-triphosphate diphosphohydrolase (4.6-fold), UDP-glucose-6-dehydrogenase (4.1-fold), and epoxide hydrolase (3.0-fold). Most of these proteins are regulated by Nrf2. However, only two proteins altered by CDDO-Me in wild-type mice were similarly affected in Nrf2−/− mice, demonstrating the high degree of selectivity of CDDO-Me for the Keap1/Nrf2 signaling pathway.
Pharmacokinetics of CDDO-Me
Due to the relatively low oral bioavailability of CDDO, new triterpenoids such as CDDO-Me with a higher oral bioavailability were synthesized. However, the oral bioavailability of unsolvated crystalline CDDO-Me was still relatively low. To improve this property, amorphous spray dried dispersion (SDD) CDDO-Me formulation has been used in the Phase III BEACON study evaluating the therapeutic effect of CDDO-Me in CKD. In a separate study, 30 mg of amorphous SDD CDDO-Me formulation showed higher bioavailability than 150 mg of the crystalline formation, while achieving a similar overall exposure profile in healthy volunteers.30
Hong et al18 developed a fast liquid chromatography/mass spectrometric method to determine the pharmacokinetics of CDDO-Me in 47 patients with advanced, refractory solid tumors and lymphoid malignancies in a Phase I trials. The most common cancer types were melanoma (16 patients), colorectal (eight patients), renal cell (five patients), anaplastic thyroid (four patients), and non-Hodgkin’s lymphoma (three patients). The main purposes of this trial were to determine the dose-limiting toxicities, maximum tolerated dose, and appropriate dose for Phase II studies; to evaluate the single-dose and steady-state pharmacokinetic parameters; and to examine the antitumor activity. SDD CDDO-Me was administered orally once daily for 21 days of a 28-day cycle. The starting dose was 5 mg, which was one-tenth the dose causing severe toxicity in rats. Blood samples were collected before dosing and at 0.25, 0.50, 1.0, 2.0, 4.0, 6.0, 8.0, and 24 hours after taking both the first and last doses during cycle 1. The pharmacokinetic behavior of the tested formulation of CDDO-Me was characterized by a slow and saturable oral absorption, a relatively long terminal phase half-life (39±20 hours at 900 mg dose level, n=19), nonlinearity (dose-dependent) at high doses (600–1,300 mg/day), and high interpatient variability.18 The median time to maximum plasma concentration was 4.1±3.4 hours at 900 mg dose level (n=19). Mean steady-state minimum and maximum concentrations of CDDO-Me in plasma from patients with daily administration of 900 mg were 8.8±4.3 ng/mL and 24.7±13.3 ng/mL, with a peak-to-trough ratio of 2.8±1.6, indicating that the once daily dosing regimen effectively maintains plasma levels of the drug within a relatively narrow range in individual patients. There was a remarkable interpatient variability in the pharmacokinetic parameters of CDDO-Me, ranging from 64% to 77% for the first dose and 39% to 54% for the last dose among patients treated at 900 mg/day. The apparent oral clearance of CDDO-Me was significantly correlated with the daily dose, whereas the terminal phase half-life was independent of the dose, suggestive of an inverse relationship between the extent of absorption and dose. A relatively long terminal phase half-life of CDDO-Me indicated that once daily dosing regimen is appropriate for future Phase II studies.
CDDO-Me for the treatment of kidney diseases
Acute kidney disease
CDDO-Me and related analogues have demonstrated therapeutic efficacy in models of acute kidney injury (AKI).31,32 Wu et al32 pretreated mice with CDDO-Me twice a day for 2 days and then subjected the mice to an ischemic model of AKI. Marked protection was noted, as manifested by an almost complete prevention of postischemic azotemia and a corresponding reduction in renal histologic injuries. CDDO-Me exerted its beneficial effect by increasing the expression of genes previously shown to protect against ischemic AKI, including Nrf2, peroxisome-activated receptor γ, and heme oxygenase-1 (HO-1). However, CDDO-Me’s effects on tubular and leukocyte HO-1 during ischemic AKI may be independent on Nrf2 pathway. It was found that CDDO-Me also ameliorated cisplatin-induced nephrotoxicity in this same study. In a recent study, CDDO-Me suppressed aristolochic acid (AA)-induced AKI via Keap1/Nrf2 pathway in male C57BL/6 mice.33 CDDO-Me at 10 mg/kg/day by ip was administered for 7 consecutive days, starting 2 days prior to aristolochic acid I (AAI) administration. CDDO-Me significantly upregulated renal Nrf2, NAD(P)H quinone oxidoreductase 1 (NQO1), and HO-1 expression and downregulated Keap1 expression compared with the AAI-treated group. Nrf2 was expressed both in the cytoplasm and nuclear of glomeruli and tubules, whereas NQO1 and HO-1 were localized in the cytoplasm of tubules only. These studies clearly demonstrate the renal protective effect of CDDO-Me via Nrf2 activation.
Chronic kidney disease
Given the role of impaired Nrf2 activity in CKD-associated oxidative stress and inflammation,34–36 CDDO-Me unexpectedly improved the renal function in patients with cancer during Phase I trials.18 Studies were designed to evaluate the effects of CDDO-Me in CKD patients with type 2 diabetes. In the first trial, 20 patients with moderate to severe CKD and type 2 diabetes were enrolled. Patients received 25 mg of CDDO-Me daily for 28 days, followed by 75 mg daily for another 28 days. Notably, there was a significant increase in estimated glomerular filtration rate (eGFR) at week 4 (+2.8 mL/min/1.73 m2) at a dose of 25 mg/day and at week 8 (+7.2 mL/min/1.73 m2) at a dose of 75 mg/day. The eGFR change paralleled a significant reduction in serum creatinine (−0.3 mg/dL) and blood urea nitrogen (BUN) (−4.9 mg/dL), along with an increase in creatinine clearance (+14.6 mL/min/1.73 m2), without a change in the 24-hour creatinine excretion rate. Markers of vascular injury and inflammation presented improvement by treatment with CDDO-Me. No life-threatening adverse events or drug-related serious adverse events were reported.37
The beneficial effect of CDDO-Me on eGFR was confirmed in a larger, multicenter, double-blind, randomized trial.38 Treatment with CDDO-Me at a target dose of 25, 75, or 150 mg once daily for 52 weeks led to sustained, significant improvements in the eGFR in patients receiving standard medical care for CKD and type 2 diabetes. A significant improvement in the primary endpoint (change of eGFR after 24 weeks postdosing) was observed in CDDO-Me-treated groups (+8.2, +11.4, and +10.4 mL/min/1.73 m2, respectively) versus 0 mL/min/1.73 m2 in the placebo group. The secondary endpoint (change of eGFR after 52 weeks postdosing) also was significantly improved in CDDO-Me-treated groups (+5.8, +10.5, and +9.3 mL/min/1.73 m2, respectively, versus 0). More patients in the placebo-treated group had an eGFR decrease more than 25% with respect to baseline value at 24 and 52 weeks postdosing. Additionally, serum BUN, phosphorus, and uric acid were significantly lower at 24 and 52 weeks postdosing in all CDDO-Me-treated groups when compared with the placebo-treated group. These results suggest that treatment with CDDO-Me may help retard disease progression in patients with advanced CKD and type 2 diabetes and justify further studies of CDDO-Me and related compounds.38 However, a higher rate of cardiovascular events with CDDO-Me than with placebo prompted termination of a Phase III trial.16 The potential reasons for the termination will be discussed later in this review.
The effect of eGFR increase and the termination of Phase III trial as mentioned previously triggered some studies to explore the mechanisms of CDDO-Me on renal protection. One of the potential mechanisms is that CDDO-Me can induce an antioxidant and anti-inflammatory response in various cells of renal origin. CDDO-Me can upregulate the expression of Nrf2 target genes such as HO-1, NQO1, and thioredoxin in mesangial cells (MCs). When these cells were exposed to albumin or tumor necrosis factor-α (TNF-α), CDDO-Me inhibited NF-κB activation and expression of cyclooxygenase 2, monocyte chemotactic protein-1, and interleukin-1β (IL-1β), providing evidence that this drug attenuates inflammation induced by factors known to be upregulated in CKD. In proximal tubular epithelial cells, glomerular endothelial cells, and podocytes, CDDO-Me was also shown to induce Nrf2 targets, as well as attenuate NF-κB activation and its downstream signaling transduction. Therefore, the main mechanism of renal preventive action of CDDO-Me was believed to be the anti-inflammatory feature that was attributed to the activation of Nrf2, inhibition of NF-κB, and decreased oxidative stress.39
Albuminuria induction effect of CDDO-Me was observed in the clinical trial. Reisman et al40 investigated whether the CDDO-Me-induced albuminuria resulted from the downregulation of megalin, a protein involved in the tubular reabsorption of albumin and lipid-bound proteins. Administration of CDDO-Me to cynomolgus monkeys significantly decreased the protein expression of megalin in renal tubules, which was inversely correlated with the urine albumin-to-creatinine ratio. Moreover, daily oral administration of CDDO-Me to monkeys for 1 year did not lead to any adverse effects on renal tissues but reduced serum creatinine and BUN, as observed in patients with CKD. The CDDO-Me-induced decrease in megalin corresponded with pharmacological induction of renal Nrf2 target genes. These findings suggest that the increase in albuminuria that accompanies CDDO-Me administration may result, at least in part, from reduced expression of megalin. This seems to occur without any adverse effects and with strong induction of Nrf2 target genes.
Since the contractile activity of MCs may influence glomerular filtration rate (GFR), Ding et al41 evaluated the effect of the synthetic triterpenoid CDDO-Me analog, RTA 405, on GFR in rats and on MC contractility in freshly isolated glomeruli. In rats, RTA 405 increased basal GFR, assessed by inulin clearance, and attenuated the angiotensin II-induced decline in GFR. RTA 405 increased the filtration fraction, but did not affect arterial blood pressure or renal plasma flow. Glomeruli from RTA 405-treated rats were resistant to angiotensin II-induced volume reduction ex vivo. In cultured MCs, angiotensin II-stimulated contraction was attenuated by RTA 405 in a dose- and time-dependent fashion. Further, Nrf2-targeted gene transcriptions in MCs were associated with decreased basal and reduced angiotensin II-stimulated hydrogen peroxide and calcium ion levels. These results implicate induction of Nrf2, reduction of intracellular reactive oxygen species levels, suppression of the glomerular MC Ca2+ response, and suppression of MC contraction as the underlying mechanisms contributing to the renal protective effect. Inhibition of MC contraction may lead to a relative increase in glomerular capillary surface area, resulting in improved GFR.
As noted previously, a higher rate of cardiovascular events with CDDO-Me than with placebo prompted termination of the Phase III trial in CKD therapy. It has been noted that the failure to the right clinical indication and heightened specificity use of CDDO-Me led to termination of the phase III trial.42 Others demonstrated that the chemical structure of CDDO-Me was similar to that of cyclopentenone prostaglandins shown to cause renal vasodilatation. It is conceivable that CDDO-Me may have increased the eGFR by causing afferent arteriolar dilatation and increasing intraglomerular pressure.43 This hemodynamic effect may result in short-term hyperfiltration, which predisposes cells to accelerated renal function loss and progression of nephropathy in the long term.44,45
CDDO-Me for the treatment of cancer
Data from the preclinical studies
In addition to its efficacy in chemical carcinogenesis models,7,46 CDDO-Me also significantly delays tumor development in various transgenic mouse models of prostate, breast, and pancreatic cancer.47–53 CDDO-Me was shown to modulate protein kinase B, NF-κB, telomerase reverse transcriptase, transcription factor signal transducers and activators of transcription 3 (STAT3), cyclin D1, human epidermal growth factor receptor 2 (EGFR2), Nrf2, and IKK signaling presenting an anticarcinogenic effect. The detailed mechanisms of this effect for CDDO-Me have been discussed in our another review recently published in Molecular Cancer.54
CDDO-Me inhibited proliferation and induced apoptosis in vitro in a wide variety of human cancer cells.8,9,11,17,55–77 It has also been shown to inhibit the growth of tumors in the mouse models.50,78,79 Although the mechanisms of the anticancer effects of CDDO-Me are not fully understood, indications from both in vitro and in vivo studies suggest that CDDO-Me can indeed modulate multiple molecular targets that play a fundamental role in both development and progression of cancer. NF-κB, phosphoinositide 3-kinase/protein kinase B1/mammalian target of rapamycin, janus-activated kinase/STAT, and death receptor-induced extrinsic apoptotic pathways are the CDDO-Me’s main targets where it performs an anticancer effect. Telomere or telomerase and immune cells of different kinds of cancers are also the targets that are modulated by CDDO-Me.
Unlike kidney diseases described previously, there is little evidence that Nrf2 activation is required for much of the anticancer activity of CDDO-Me. The most possible reason is that CDDO-Me targets the cysteine residues of multiple proteins in a dose-dependent manner. As mentioned previously, drugs that undergo Michael addition have different binding affinities for different target proteins. Low concentrations of drug might preferentially interact with targets such as Keap1 to release Nrf2 and activate the phase 2 cytoprotective pathway, whereas higher concentrations of triterpenoids might interact with target proteins with lower binding affinities such as tubulin or IKK to inhibit proliferation and induce apoptosis.14 Another reason for a low Nrf2 dependence by CDDO-Me is that cancer cells can hijack the Keap1/Nrf2/ARE system, acquiring malignant properties. Several mechanisms have been reported for the hijack effect of Keap1/Nrf2/ARE pathway: somatic mutations in Keap1 or Nrf2, deoxyribonucleic acid (DNA) hypermethylation at the promoter region of Keap1, the aberrant accumulation of proteins that disrupt the Keap1/Nrf2 interaction, transcriptional upregulation of the Nrf2 gene through oncogene-dependent signaling, and the modification of Keap1 protein through oncometabolites. The hijack effect can make CDDO-Me lose its Nrf2 activation function.80 Lastly, benign cells are under much greater control from inflammatory cells and other stromal cells in their microenvironment and, moreover, they have not yet reached a level of DNA damage that makes them autonomous. Therefore, enhancement of Nrf2 activity appears more obvious in benign states than in the malignant states.15
Data from the clinical trials
CDDO-Me shows promise in preventive and therapeutic activities against several types of cancer not only in both in vitro and animal studies, but also in clinical trials.14,54,81,82 Nagaraj et al17 analyzed samples from 19 patients with pancreatic adenocarcinoma that were treated with CDDO-Me in the Phase I clinical trial RTA 402-C-0702. Patients were treated intravenously with gemcitabine (1,000 mg/m2) weekly on days 1, 8, and 15. CDDO-Me was administered orally once daily for 21 days. Nine patients received a dose of 150 mg/day; two patients, 200 mg/day; six patients, 250 mg/day; and two patients, 300 mg/day. CDDO-Me abrogated the immune suppressive effect of myeloid-derived suppressor cells (MDSCs) and improved immune responses in cancer patients. No toxicity attributed to CDDO-Me was observed.
Hong et al18 reported the data of Phase I clinical trial of CDDO-Me in 47 patients with advanced solid tumors and lymphoma. They found that CDDO-Me-induced dose limited toxicity was reversible and exhibited grade 3 liver transaminase elevation. The maximum tolerated dose was established as 900 mg/day. A complete tumor response occurred in a mantle cell lymphoma patient, and a partial response was observed in an anaplastic thyroid carcinoma patient. NQO1 messenger ribonucleic acid (mRNA) levels increased in peripheral blood mononuclear cells, and NF-κB and cyclin D1 levels decreased in tumor biopsies. They concluded that CDDO-Me was well tolerated with a maximum tolerated dose of 900 mg/day. For the observed objective tumor responses, CDDO-Me was suggested for further development in solid cancer (Table 1).
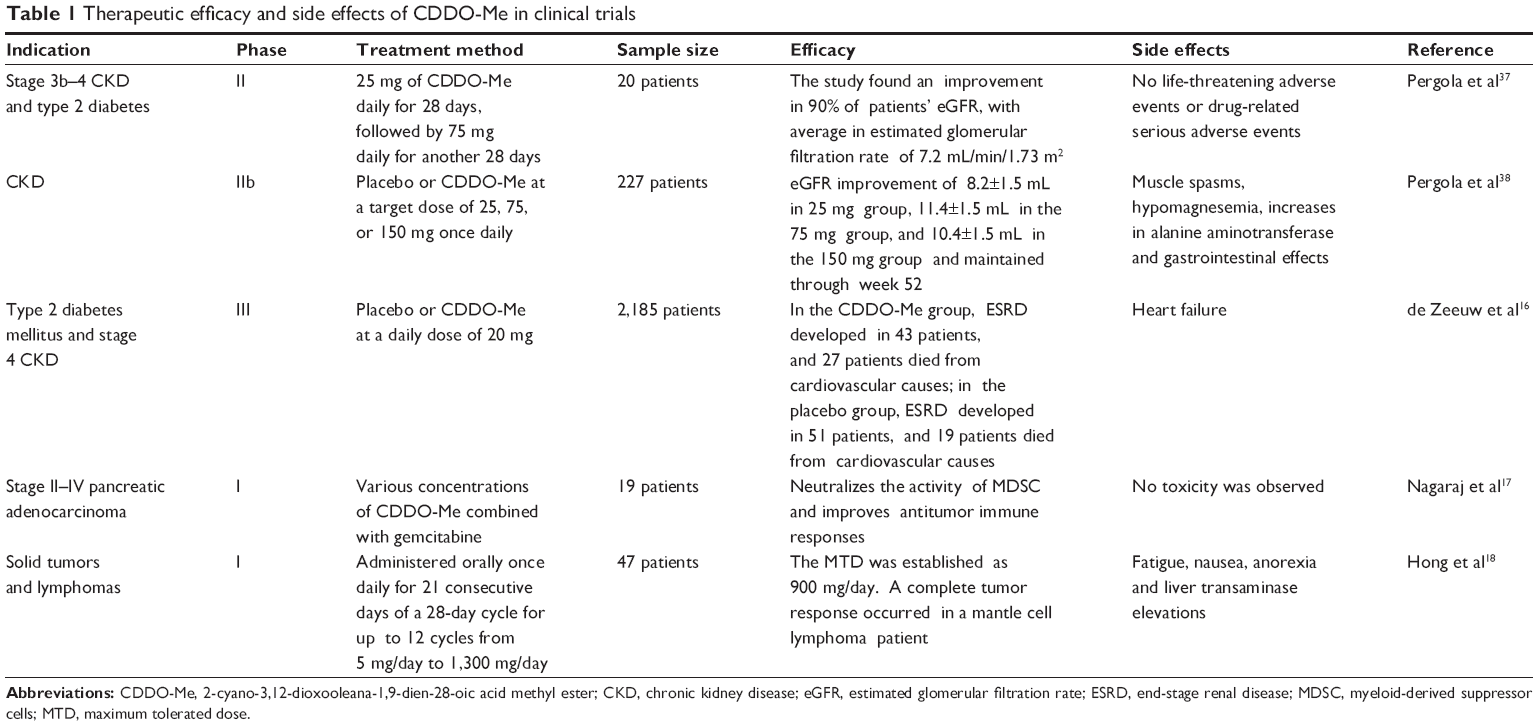 | Table 1 Therapeutic efficacy and side effects of CDDO-Me in clinical trials |
Treatment of other diseases by CDDO-Me
Besides kidney disease and malignant diseases, activation of the Nrf2/ARE and inhibition of NF-κB pathway by CDDO-Me should protect against a number of other diseases driven by inflammation and oxidative stress (Table 2).
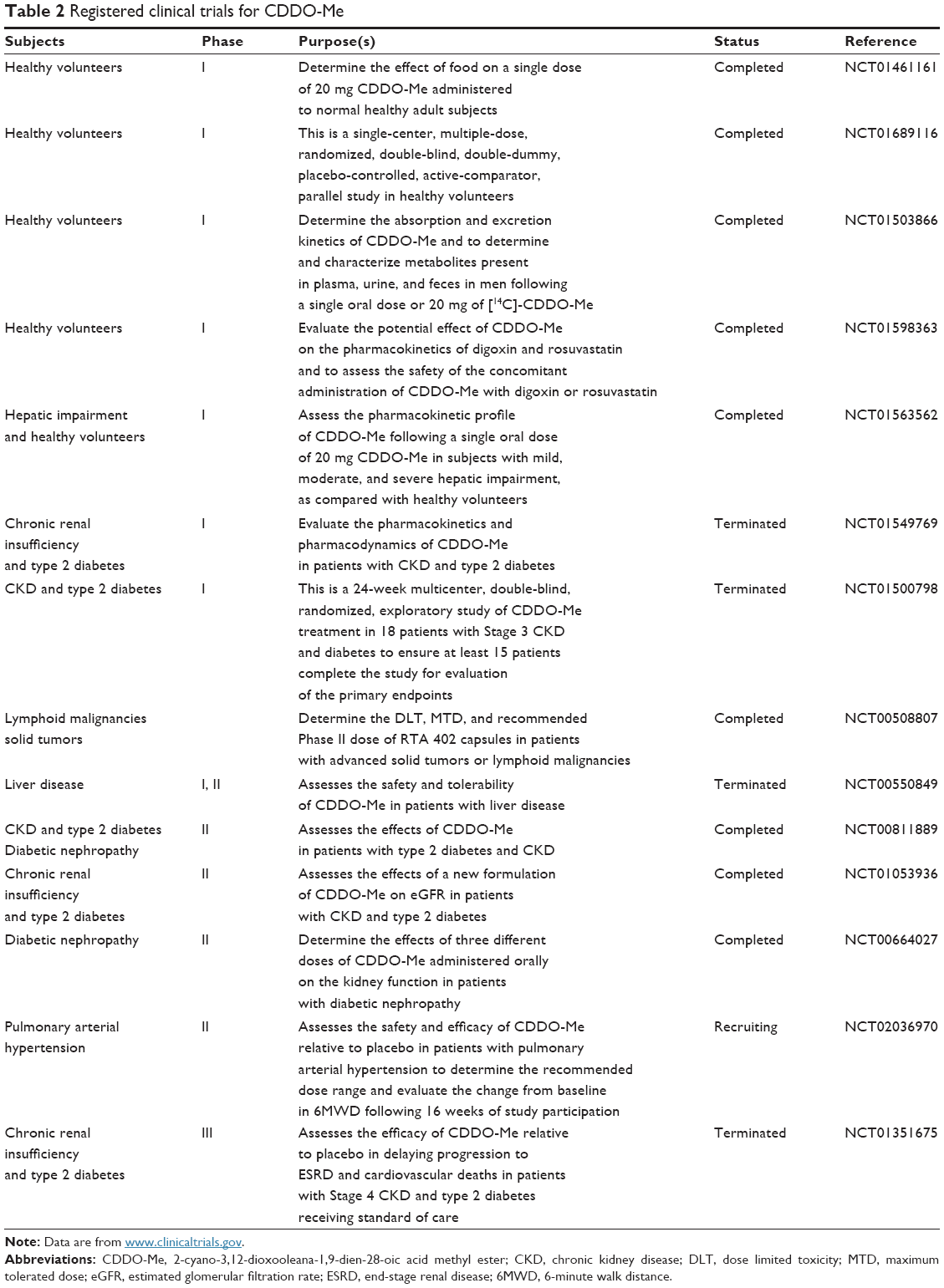 | Table 2 Registered clinical trials for CDDO-Me |
Neuroprotective effect
Tran et al83 found that at low nanomolar concentrations, treatment of rat primary mesencephalon neuron/glia cultures with CDDO-Me resulted in attenuated lipopolysaccharide (LPS)-, TNF- or fibrillar β-amyloid 1–42 peptide-induced increases in reactive microglia and inflammatory gene expression without an overall effect on cell viability. In addition, CDDO-Me blocked death in the dopaminergic neuron-like cell line MN9D induced by conditioned media of LPS-stimulated BV2 microglia, but did not block cell death induced by addition of TNF-α to MN9D cells, suggesting that dopaminergic neuroprotection by CDDO-Me involved inhibition of microglia-derived cytokine production and not direct inhibition of TNF-α-dependent proapoptotic pathways. The authors then found that the inhibition of reactive oxygen species and specific subsets of cytokines by CDDO-Me may be the underlying mechanisms for the neuroprotection effect. The neuroimmune modulatory properties of CDDO-Me indicate that this potent antioxidant and anti-inflammatory compound may have a therapeutic potential to modify the course of neurodegenerative diseases such as Alzheimer’s disease characterized by chronic neuroinflammation and amyloid deposition.
Radioprotective effect
The activation of the Nrf2/ARE pathway by CDDO-Me protects cells from a variety of chemical or physical insults. Kim et al84 reported that CDDO-Me protected colonic epithelial cells against radiation-induced damage, in part, by enhancing signaling of the DNA damage response. Pretreatment with CDDO-Me reduced the frequency of both G1 and S/G2 chromosome aberrations and enhanced the disappearance of repairosomes (C-terminal binding protein interacting protein, Rad51, and p53 binding protein-1 foci) after irradiation. CDDO-Me protected cells from radiation toxicity in an Nrf2-dependent manner. The p53 binding protein-1 promoter contains three antioxidant responsive elements in which Nrf2 directly binds following CDDO-Me treatment. These results demonstrate that Nrf2 activation by synthetic triterpenoids is a promising candidate target to protect the gastrointestinal tract against acute irradiation. Eskiocak et al85 found that CDDO-Me was able to activate the Nrf2/ARE pathway and protect human colon epithelial cells against radiation-induced DNA transformation. Pretreatment of cells with CDDO-Me prevented the heavy-ion-induced increase in proliferation rate and anchorage-dependent and -independent colony formation efficiencies.
However, an Nrf2-independent radiation protective effect of CDDO-Me was also observed in some studies. Kim et al86 investigated whether CDDO-Me mitigated radiation-induced DNA damage in immortalized normal human colonic epithelial cells and bronchial epithelial cells. DNA damage and clonogenic survival were assessed after treatment with CDDO-Me postirradiation. They observed that treatment with CDDO-Me within 30 minutes after irradiation improved both DNA damage repair and clonogenic survival. CDDO-Me activates EGFR-related DNA repair responses. In the presence of CDDO-Me, EGFR is phosphorylated and translocates into the nucleus where it interacts with DNA-dependent protein kinase, catalytic subunit (DNA-PKcs). CDDO-Me-mediated mitigation activity can be abrogated through depletion of EGFR, ectopic overexpression of mutant EGFR, or inhibition of DNA-PKcs, which is independent of Nrf2.
Antifibrotic effect
The CDDO-Me also provides protection against radiation- or chemical-induced fibrosis of different organs. Kulkarni et al87 explored whether CDDO-Me could reduce lung inflammation, fibrosis, and lung function impairment in a bleomycin-induced lung injury and fibrosis model. They found that CDDO-Me had broad anti-inflammatory effects against multiple cytokines, such as transforming growth factor-β (TGF-β) and IL-6, which are involved in the initiation and development of fibrosis. They indicated that early administration of CDDO-Me potently inhibited inflammation, reduced fibrotic outcomes, and significantly improved lung function in bleomycin-treated mice.
Wounds to the cornea, the second most common cause of blindness, initiate the release of cytokines such as TGF-β that induce keratocytes to differentiate into myofibroblasts. Kuriyan et al88 reported that CDDO-Me could function as a potent antifibrotic agent for cultured human corneal fibroblasts stimulated to differentiate into myofibroblasts by TGF-β. CDDO-Me inhibited the differentiation of primary corneal fibroblasts treated with TGF-β into myofibroblasts, as measured by the complete suppression of the in vitro production of α-smooth muscle actin, collagen, and fibronectin. Reactivity with intracellular nucleophiles may be a potential mechanism through which CDDO-Me was able to inhibit myofibroblast differentiation.
Inflammatory/metabolic disorders
Auletta et al89 found that pretreatment with oral CDDO-Me significantly improved survival following lethal-dose LPS challenge in mice. CDDO-Me extends the survival of mice challenged with LPS and decreases circulating levels of IL-6, IL-12, IL-17, IL-23, and interferons without altering the numbers or subtypes of immune cells.
Saha et al90 reported that CDDO-Me ameliorated diabetes in mice fed a high-fat diet or in mice with a defective leptin receptor (Leprdb/db). Oral CDDO-Me administration reduced total body fat, plasma triglyceride, and free fatty acid levels. It also improved glucose and insulin tolerance tests. Although CDDO-Me reduces total body fat and suppresses production of the proinflammatory cytokines IL-1, IL-6, and TNF-α in mice fed a high-fat diet, the antidiabetic effects of this agent may be mediated by stimulating phosphorylation of liver kinase B1 and AMP-activated protein kinase (AMPK) in muscle and liver, as knockdown of AMPK reduces glucose uptake in cells treated with CDDO-Me.
Pulmonary arterial hypertension
Pulmonary arterial hypertension is a life-threatening disease involving endothelial dysfunction, vasoconstriction in small pulmonary arteries, aberrant proliferation of certain vascular cells, and dysregulated inflammatory signaling leading to vascular remodeling, pulmonary fibrosis, and right ventricular hypertrophy.91 Preclinical data have demonstrated that CDDO-Me could modulate the endothelin (ET) pathway and reduce vascular inflammation. Chin et al92 found CDDO-Me administration was associated with decreases in ET-1 secretion and ET receptors type A (ETA) receptor expression. Furthermore, the ET pathway has been identified as a target of NF-κB, with ETA expression decreased in renal artery organ cultures treated with direct NF-κB inhibitors.93 Thus, the inhibitory effects of CDDO-Me on the NF-κB pathway may contribute to the reduced activity of ET pathway. In addition, CDDO-Me and related compounds have highly potent antioxidant, anti-inflammatory properties, as well as antiproliferative and antifibrotic effects, which has been discussed in the previous part of this review. On the basis of these data, CDDO-Me is currently being studied in patients with pulmonary arterial hypertension in a Phase I trial.
Graft-versus-host disease
Although the mechanisms responsible for treatment of graft-versus-host disease (GVHD) by the CDDO-Me are not completely understood, several clues have emerged. Ames et al94 observed that CDDO-Me promoted myelopoiesis in both naïve and transplanted mice. After sublethal total body irradiation, mice pretreated with CDDO-Me further displayed an accelerated recovery of myeloid progenitors and total nucleated cells in the spleen. A similar expansion of myeloid and myeloid progenitors was noted with CDDO-Me treatment after syngeneic bone marrow transplantation. Compared with granulocyte colony-stimulating factor, CDDO-Me not only expanded CD11b+ Gr-1+ myeloid cells in the bone marrow and spleen, but also increased the granulocyte macrophage-colony forming unit colonies in the spleen, bone marrow, and blood. Additionally, the number of granulocyte macrophage-colony forming units and hematopoietic progenitor cells in the spleen and bone marrow showed a significant increase in mice that received CDDO-Me. In vitro studies also showed that CDDO-Me was sufficient to stimulate colony growth of naïve bone marrow cells in suboptimal colony-stimulating cytokine conditions. These results suggest that CDDO-Me may be of use posttransplantation to accelerate myeloid recovery in addition to the prevention of GVHD. Li et al95 found that CDDO-Me had an increased ability to inhibit allogeneic T cell responses and induce cell death of alloreactive T cells in vitro. Administration of CDDO-Me to mice following allogeneic bone marrow transplantation resulted in significant and increased protection from lethal acute GVHD compared with CDDO. This was associated with reduced donor T cell proliferation, decreased adhesion molecule (α4β7 integrin) expression on the donor T cells, and reduced production of the proinflammatory cytokine TNF-α. These results suggest that CDDO-Me may be of significant benefit in suppressing acute GVHD, while preserving or possibly even augmenting graft-versus-tumor effects if it is applied together with allogeneic bone marrow transplantation in cancer.
Conclusion and future perspective
Inflammation and oxidative stress are hallmarks and mediators of the progression of most diseases. Previous studies with CDDO-Me have demonstrated that this compound had the capability of modifying a wide variety of proteins through nucleophilic attack and Michael addition, particularly at vulnerable -SH groups. The specific structure supports CDDO-Me to upregulate the antioxidant response, while diminishing proinflammatory signaling. Moreover, because large collections of regulatory proteins have reactive -SH groups, the targets of CDDO-Me are multiple, and the interaction between CDDO-Me and targets may result in a broad collection of direct and indirect effects. As a multifunctional agent, the therapeutic effects of CDDO-Me have been tested in clinical trials for CKD and malignant diseases. The potential role of CDDO-Me in treatment of other diseases is being explored in both in vivo and in vitro models. In the future, the underlying mechanisms of therapeutic effects for CDDO-Me should be explored further to identify new indications for this multifunctional drug. In addition, the combined use of CDDO-Me with other agents or targeted therapy should be examined. The dose–response and dose–toxicity relationships for CDDOM-Me should be established to optimize its dosing regimens. The data on its disposition, metabolism, and transport are lacking. Finally, although the agent appears to be well tolerated and side effects appear to be largely mild, transient, and self-limiting, additional safety data are eagerly awaited.
Disclosure
The authors report no conflicts of interest in this work.
References
Huang MT, Ho CT, Wang ZY, et al. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994;54(3):701–708. | ||
Nishino H, Nishino A, Takayasu J, et al. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-acetate by some oleanane-type triterpenoid compounds. Cancer Res. 1988;48(18):5210–5215. | ||
Honda T, Rounds BV, Gribble GW, Suh NJ, Wang YP, Sporn MB. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett. 1998;8(19):2711–2714. | ||
Suh NJ, Wang YP, Honda T, et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59(2):336–341. | ||
Konopleva M, Tsao T, Estrov Z, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64(21):7927–7935. | ||
Honda T, Rounds BV, Bore L, et al. Synthetic oleanane and ursane triterpenoids with modified rings A and C: A series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem. 2000;43(22):4233–4246. | ||
Liby K, Royce DB, Williams CR, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67(6):2414–2419. | ||
Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3, 12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor-γ-dependent and -independent pathways. Mol Pharmacol. 2005;68(1):119–128. | ||
Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99(1):326–335. | ||
Ikeda T, Sporn M, Honda T, Gribble GW, Kufe D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63(17):5551–5558. | ||
Hyer ML, Shi R, Krajewska M, et al. Apoptotic activity and mechanism of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic-acid and related synthetic triterpenoids in prostate cancer. Cancer Res. 2008;68(8):2927–2933. | ||
Yates MS, Tauchi M, Katsuoka F, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6(1):154–162. | ||
Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005;102(12):4584–4589. | ||
Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7(5):357–369. | ||
Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64(4):972–1003. | ||
de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369(26):2492–2503. | ||
Nagaraj S, Youn JI, Weber H, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16(6):1812–1823. | ||
Hong DS, Kurzrock R, Supko JG, et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2012;18(12):3396–3406. | ||
Sporn MB, Liby KT, Yore MM, Fu LF, Lopchuk JM, Gribble GW. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod. 2011;74(3):537–545. | ||
Pollier J, Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. | ||
Couch RD, Browning RG, Honda T, et al. Studies on the reactivity of CDDO, a promising new chemopreventive and chemotherapeutic agent: implications for a molecular mechanism of action. Bioorg Med Chem Lett. 2005;15(9):2215–2219. | ||
Yore MM, Kettenbach AN, Sporn MB, Gerber SA, Liby KT. Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS One. 2011;6(7):e22862. | ||
Wang YP, Porter WW, Suh NJ, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor-γ. Mol Endocrinol. 2000;14(10):1550–1556. | ||
Kawamoto Y, Nakamura Y, Naito Y, et al. Cyclopentenone prostaglandins as potential inducers of phase II detoxification enzymes. 15-deoxy-Δ12,14-prostaglandin J2-induced expression of glutathione S-transferases. J Biol Chem. 2000;275(15):11291–11299. | ||
Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403(6765):103–108. | ||
Levonen AL, Landar A, Ramachandran A, et al. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378(pt 2):373–382. | ||
Straus DS, Pascual G, Li M, et al. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci U S A. 2000;97(9):4844–4849. | ||
Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-κB pathway by direct inhibition of IKKβ on Cys-179. J Biol Chem. 2006;281(47):35764–35769. | ||
Walsh J, Jenkins RE, Wong M, et al. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: Biochemical, pharmacological and toxicological implications. J Proteomics. 2014;108:171–187. | ||
Thomas M. A preliminary evaluation of bardoxolone methyl for the treatment of diabetic nephropathy. Expert Opin Drug Metab Toxicol. 2012;8(8):1015–1022. | ||
Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, Klaassen CD. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J Pharmacol Exp Ther. 2010;335(1):2–12. | ||
Wu QQ, Wang Y, Senitko M, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol Renal Physiol. 2011;300(5):F1180–F1192. | ||
Wu J, Liu X, Fan J, et al. Bardoxolone methyl (BARD) ameliorates aristolochic acid (AA)-induced acute kidney injury through Nrf2 pathway. Toxicology. 2014;318:22–31. | ||
Yoh K, Hirayama A, Ishizaki K, et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes to Cells. 2008;13(11):1159–1170. | ||
Yoh K, Itoh K, Enomoto A, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60(4):1343–1353. | ||
Liu M, Grigoryev DN, Crow MT, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76(3):277–285. | ||
Pergola PE, Krauth M, Huff JW, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b–4 CKD. Am J Nephrol. 2011;33(5):469–476. | ||
Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365(4):327–336. | ||
Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83(6):1029–1041. | ||
Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ. Bardoxolone methyl decreases megalin and activates Nrf2 in the kidney. J Am Soc Nephrol. 2012;23(10):1663–1673. | ||
Ding YF, Stidham RD, Bumeister R, et al. The synthetic triterpenoid, RTA 405, increases the glomerular filtration rate and reduces angiotensin II-induced contraction of glomerular mesangial cells. Kidney Int. 2013;83(5):845–854. | ||
Zhang DD. Bardoxolone brings Nrf2-based therapies to light. Antioxid Redox Signal. 2013;19(5):517–518. | ||
Tayek JA, Kalantar-Zadeh K. The extinguished BEACON of bardoxolone: not a Monday morning quarterback story. Am J Nephrol. 2013;37(3):208–211. | ||
Ruggenenti P, Porrini EL, Gaspari F, et al. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care. 2012;35(10):2061–2068. | ||
Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dialysis Transplant. 2014;29(suppl 1):i19–i24. | ||
Liby KT. Synthetic triterpenoids can protect against toxicity without reducing the efficacy of treatment with carboplatin and paclitaxel in experimental lung cancer. Dose Response. 2014;12(1):136–151. | ||
Deeb D, Gao XH, Liu YB, et al. Synthetic triterpenoid CDDO prevents the progression and metastasis of prostate cancer in TRAMP mice by inhibiting survival signaling. Carcinogenesis. 2011;32(5):757–764. | ||
Gao X, Deeb D, Liu Y, et al. Prevention of prostate cancer with oleanane synthetic triterpenoid CDDO-Me in the TRAMP mouse model of prostate cancer. Cancers (Basel). 2011;3(3):3353–3369. | ||
Liu YB, Gao XH, Deeb D, Arbab AS, Gautam SC. Telomerase reverse transcriptase (TERT) is a therapeutic target of oleanane triterpenoid CDDO-Me in prostate cancer. Molecules. 2012;17(12):14795–14809. | ||
Liby K, Risingsong R, Royce DB, et al. Prevention and treatment of experimental estrogen receptor-negative mammary carcinogenesis by the synthetic triterpenoid CDDO-methyl ester and the rexinoid LG100268. Clin Cancer Res. 2008;14(14):4556–4563. | ||
Tran K, Risingsong R, Royce D, Williams CR, Sporn MB, Liby K. The synthetic triterpenoid CDDO-methyl ester delays estrogen receptor-negative mammary carcinogenesis in polyoma middle T mice. Cancer Prev Res. 2012;5(5):726–734. | ||
Kim EH, Deng CX, Sporn MB, et al. CDDO-methyl ester delays breast cancer development in Brca1-mutated mice. Cancer Prev Res. 2012;5(1):89–97. | ||
Liby KT, Royce DB, Risingsong R, et al. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev Res. 2010;3(11):1427–1434. | ||
Wang YY, Zhe H, Zhao R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol Cancer. 2014;13:30. | ||
Deeb D, Gao X, Liu Y, Varma NR, Arbab AS, Gautam SC. Inhibition of telomerase activity by oleanane triterpenoid CDDO-Me in pancreatic cancer cells is ROS-dependent. Molecules. 2013;18(3):3250–3265. | ||
Gao X, Liu Y, Deeb D, et al. ROS mediate proapoptotic and antisurvival activity of oleanane triterpenoid CDDO-Me in ovarian cancer cells. Anticancer Res. 2013;33(1):215–221. | ||
Qin Y, Deng W, Ekmekcioglu S, Grimm EA. Identification of unique sensitizing targets for anti-inflammatory CDDO-Me in metastatic melanoma by a large-scale synthetic lethal RNAi screening. Pigment Cell Melanoma Res. 2013;26(1):97–112. | ||
Deeb D, Gao X, Liu Y, et al. Inhibition of cell proliferation and induction of apoptosis by oleanane triterpenoid (CDDO-Me) in pancreatic cancer cells is associated with the suppression of hTERT gene expression and its telomerase activity. Biochem Biophys Res Commun. 2012;422(4):561–567. | ||
Liu Y, Gao X, Deeb D, Gautam SC. Oleanane triterpenoid CDDO-Me inhibits Akt activity without affecting PDK1 kinase or PP2A phosphatase activity in cancer cells. Biochem Biophys Res Commun. 2012;417(1):570–575. | ||
Gao X, Liu Y, Deeb D, et al. Synthetic oleanane triterpenoid, CDDO-Me, induces apoptosis in ovarian cancer cells by inhibiting prosurvival AKT/NF-kappaB/mTOR signaling. Anticancer Res. 2011;31(11):3673–3681. | ||
Deeb D, Gao X, Arbab AS, Barton K, Dulchavsky SA, Gautam SC. CDDO-Me: A novel synthetic triterpenoid for the treatment of pancreatic cancer. Cancers (Basel). 2010;2(4):1779–1793. | ||
Ahmad R, Liu S, Weisberg E, et al. Combining the FLT3 inhibitor PKC412 and the triterpenoid CDDO-Me synergistically induces apoptosis in acute myeloid leukemia with the internal tandem duplication mutation. Mol Cancer Res. 2010;8(7):986–993. | ||
Ryu K, Susa M, Choy E, et al. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC Cancer. 2010;10:187. | ||
Yang C, Hornicek FJ, Wood KB, et al. Blockage of Stat3 with CDDO-Me inhibits tumor cell growth in chordoma. Spine (Phila Pa 1976). 2010;35(18):1668–1675. | ||
Deeb D, Gao X, Jiang H, Dulchavsky SA, Gautam SC. Oleanane triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells by independently targeting pro-survival Akt and mTOR. Prostate. 2009;69(8):851–860. | ||
Zou W, Yue P, Khuri FR, Sun SY. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008;68(18):7484–7492. | ||
Duan Z, Ames RY, Ryan M, Hornicek FJ, Mankin H, Seiden MV. CDDO-Me, a synthetic triterpenoid, inhibits expression of IL-6 and Stat3 phosphorylation in multi-drug resistant ovarian cancer cells. Cancer Chemother Pharmacol. 2009;63(4):681–689. | ||
Zou W, Chen S, Liu X, et al. c-FLIP downregulation contributes to apoptosis induction by the novel synthetic triterpenoid methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate (CDDO-Me) in human lung cancer cells. Cancer Biol Ther. 2007;6(10):1614–1620. | ||
Deeb D, Gao X, Dulchavsky SA, Gautam SC. CDDO-Me induces apoptosis and inhibits Akt, mTOR and NF-κB signaling proteins in prostate cancer cells. Anticancer Res. 2007;27(5A):3035–3044. | ||
Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IκBα kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor κB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12(6):1828–1838. | ||
Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1) → signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008;68(8):2920–2926. | ||
Zou W, Liu XG, Yue P, et al. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res. 2004;64(20):7570–7578. | ||
Gao X, Deeb D, Hao J, et al. Synthetic triterpenoids inhibit growth, induce apoptosis and suppress pro-survival Akt, mTOR and NF-κB signaling proteins in colorectal cancer cells. Anticancer Res. 2010;30(3):785–792. | ||
Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40(11):2969–2975. | ||
Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA, Gautam SC. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-κB and Notch1 signaling. J Neurooncol. 2007;84(2):147–157. | ||
Samudio I, Kurinna S, Ruvolo P, et al. Inhibition of mitochondrial metabolism by methyl-2-cyano-3,12-dioxooleana-1,9-diene-28-oate induces apoptotic or autophagic cell death in chronic myeloid leukemia cells. Mol Cancer Ther. 2008;7(5):1130–1139. | ||
Samudio I, Konopleva M, Pelicano H, et al. A novel mechanism of action of methyl-2-cyano-3,12 dioxoolean-1,9 diene-28-oate: direct permeabilization of the inner mitochondrial membrane to inhibit electron transport and induce apoptosis. Mol Pharmacol. 2006;69(4):1182–1193. | ||
Liby K, Risingsong R, Royce DB, et al. Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev Res. 2009;2(12):1050–1058. | ||
Jutooru I, Chadalapaka G, Abdelrahim M, et al. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol Pharmacol. 2010;78(2):226–236. | ||
Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:200. | ||
Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drug. 2009;20(10):880–892. | ||
Shanmugam MK, Dai XY, Kumar AP, Tan BKH, Sethi G, Bishayee A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: preclinical and clinical evidence. Cancer Lett. 2014;346(2):206–216. | ||
Tran TA, McCoy MK, Sporn MB, Tansey MG. The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection. J Neuroinflammation. 2008;5:14. | ||
Kim SB, Pandita RK, Eskiocak U, et al. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc Natl Acad Sci U S A. 2012;109(43):E2949–E2955. | ||
Eskiocak U, Kim SB, Roig AI, et al. CDDO-Me protects against space radiation-induced transformation of human colon epithelial cells. Radiat Res. 2010;174(1):27–36. | ||
Kim SB, Ly P, Kaisani A, Zhang L, Wright WE, Shay JW. Mitigation of radiation-induced damage by targeting EGFR in noncancerous human epithelial cells. Radiat Res. 2013;180(3):259–267. | ||
Kulkarni AA, Thatcher TH, Hsiao HM, et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One. 2013;8(5):e63798. | ||
Kuriyan AE, Lehmann GM, Kulkarni AA, et al. Electrophilic PPARγ ligands inhibit corneal fibroblast to myofibroblast differentiation in vitro: a potentially novel therapy for corneal scarring. Exp Eye Res. 2012;94(1):136–145. | ||
Auletta JJ, Alabran JL, Kim BG, Meyer CJ, Letterio JJ. The synthetic triterpenoid, CDDO-Me, modulates the proinflammatory response to in vivo lipopolysaccharide challenge. J Interferon Cytokine Res. 2010;30(7):497–508. | ||
Saha PK, Reddy VT, Konopleva M, Andreeff M, Chan L. The triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic-acid methyl ester has potent anti-diabetic effects in diet-induced diabetic mice and Leprdb/db mice. J Biolog Chem. 2010;285(52):40581–40592. | ||
D’Alto M, Diller GP. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: current advanced management strategies. Heart. 2014;100(17):1322–1328. | ||
Chin MP, Reisman SA, Bakris GL, et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am J Nephrol. 2014;39(6):499–508. | ||
Xie YH, Wang SW, Zhang Y, Edvinsson L, Xu CB. Nuclear factor-κB-mediated endothelin receptor up-regulation increases renal artery contractility in rats. Basic Clin Pharmacol Toxicol. Epub 2013 Aug 22. | ||
Ames E, Harouna S, Meyer C, Welniak LA, Murphy WJ. The triterpenoid CDDO-Me promotes hematopoietic progenitor expansion and myelopoiesis in mice. Biol Blood Marrow Transplant. 2012;18(3):396–405. | ||
Li M, Sun K, Redelman D, Welniak LA, Murphy WJ. The triterpenoid CDDO-Me delays murine acute graft-versus-host disease with the preservation of graft-versus-tumor effects after allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2010;16(6):739–750. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.