")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 7
Pharmacokinetic equivalence study of two formulations of the anticonvulsant pregabalin
Authors Tjandrawinata R , Setiawati E, Putri R, Gunawan V, Ong F, Susanto L , Nofiarny D
Received 3 February 2015
Accepted for publication 3 March 2015
Published 20 April 2015 Volume 2015:7 Pages 69—75
DOI https://doi.org/10.2147/CPAA.S82143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Arthur E. Frankel
Video abstract presented by Raymond R Tjandrawinata
Views: 1603
Raymond R Tjandrawinata,1 Effi Setiawati,2 Ratih Sofia Ika Putri,2 Vincent Angga Gunawan,2 Fenny Ong,1 Liana W Susanto,1 Dwi Nofiarny1
1Dexa Laboratories of Biomolecular Sciences, Cikarang, West Java, Indonesia; 2PT Equilab International Bioavailability and Bioequivalence Laboratory, Jakarta, Indonesia
Purpose: The present study was conducted to evaluate whether the bioavailability of pregabalin capsules 150 mg manufactured by PT Dexa Medica was equivalent to the reference formulation.
Methods: This was a randomized, open-label, two-period, two-sequence, and crossover study under fasting condition, with a 1-week washout period. Plasma concentrations of pregabalin from 20 subjects were determined by using a validated liquid chromatography with tandem mass spectrometry (LC-MS/MS) detection method. Pharmacokinetic parameters assessed in this study were: area under the plasma concentration–time curve from time zero to last observed quantifiable concentration (AUC0–t), area under the plasma concentration–time curve from time zero to infinity (AUC0–∞), maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), and terminal half-life (t1/2). The 90% confidence intervals (CIs) for the geometric mean ratios of test formulation/reference formulation were calculated for the AUC and Cmax parameters; while tmax difference was analyzed nonparametrically on the original data using the Wilcoxon matched-pairs test, and t1/2 difference was analyzed using Student's paired t-test.
Results: The mean (standard deviation [SD]) AUC0–t, AUC0–∞, Cmax, and t1/2 of pregabalin from the test formulation were 27,845.86 (4,508.27) ng·h/mL, 28,311.70 (4,790.55) ng·h/mL, 3,999.71 (801.52) ng/mL, and 5.66 (1.20) hours, respectively; while the mean (SD) AUC0–t, AUC0–∞, Cmax, and t1/2 of pregabalin from the reference formulation were 27,398.12 (4,266.28) ng·h/mL, 27,904.24 (4,507.31) ng·h/mL, 3,849.50 (814.50) ng/mL, and 5.87 (1.25) hours, respectively. The median (range) tmax of pregabalin from the test formulation and reference formulation was 1.00 (0.67–2.00) hours and 1.00 (0.67–3.00) hours, respectively. The 90% CIs for the geometric mean ratios of test formulation/reference formulation for pregabalin were 101.54% (98.75%–104.41%) for AUC0–t, 101.35% (98.66%–104.11%) for AUC0–∞, and 104.19% (98.75%–109.93%) for Cmax.
Conclusion: The study concluded that the two formulations of pregabalin capsules studied were bioequivalent.
Keywords: antiepileptic, bioavailability, bioequivalence, generic product
Introduction
Pregabalin is described chemically as (S)-3-(aminomethyl)-5-methylhexanoic acid, with the molecular formula C8H17NO2 and molecular weight of 159.23 Da. The chemical structure of pregabalin is shown in Figure 1. Pregabalin is considered to be highly soluble, rapidly dissolves in water, basic, or acidic aqueous solutions, and is highly permeable.1
Pregabalin is used in combination with other anticonvulsant agents in the treatment of partial seizures. It is also indicated for management of neuropathic pain, including postherpetic neuralgia and pain associated with diabetic peripheral neuropathy, as well as generalized anxiety disorder and fibromyalgia. The dose ranges from 150 to 600 mg/day and is given in either two or three divided doses. Pregabalin binds with high affinity to an auxiliary subunit (α2-δ protein) of voltage-gated calcium channels in the central nervous system (CNS), which is responsible for its analgesic and anticonvulsant effects. Pregabalin reduces calcium-dependent release of several neurotransmitters by the modulation of calcium channel function. It is structurally related to the inhibitory CNS neurotransmitter γ-aminobutyric acid (GABA). However, the drug does not bind directly to GABAA, GABAB, or benzodiazepine receptors, does not increase GABAA response in cultured neurons, and does not alter brain concentrations of GABA or affect GABA uptake or degradation.2–4
 | Figure 1 Chemical structure of pregabalin. |
Pregabalin pharmacokinetics are dose proportional over the range of recommended daily doses. It also has low intersubject pharmacokinetic variability – according to Bockbrader et al,5 the intersubject variability of pregabalin, reported as coefficient of variation (CV), is below 15%. Pregabalin is rapidly absorbed after oral doses and takes approximately 1.5 hours to achieve its maximum plasma concentrations (Cmax). Its oral bioavailability is more than 90% and is independent of dose. The rate of pregabalin absorption is reduced when given with food, resulting in an approximate 25% to 30% decrease in Cmax and an approximate 3-hour increase in time to Cmax (tmax), but this is not clinically significant, as the extent of absorption is not affected. Therefore, it can be taken with or without food. Pregabalin is not bound to plasma proteins and has been shown to cross the blood–brain barrier as well as the placenta in preclinical studies.6 Pregabalin undergoes insignificant metabolism in humans. More than 90% of the dose is excreted in urine as unchanged drug, with a terminal half-life (t1/2) of 4.8 to 6.3 hours in subjects with normal renal function. Race and sex do not significantly affect the pharmacokinetics of pregabalin, but the dosing of this drug has to be adjusted according to creatinine clearance. Pregabalin does not interact with other drugs because of its lack of hepatic metabolism and interaction with cytochrome P-450 isoenzymes.1,3,4
According to World Health Organization (WHO) estimation, eight in one thousand people in the world have epilepsy.7 The prevalence in developing countries is even higher. More than half of the total epilepsy patients worldwide are estimated to live in Asia.8–10 The condition results in a vast need of antiepileptic drugs, such as pregabalin. However, availability of the drug has become one of the most prevailing problems in the disease management.11 Therefore generic products are urgently needed to satisfy patients’ needs. Not only having to provide availability in the market and offer more competitive pricing, generic products also have to demonstrate therapeutic equivalence to the reference (innovator’s) product. Therapeutic equivalence is defined as the sharing of the same beneficial and adverse effects between two products or formulations, resulting in the interchangeability of both products.12,13
To ensure therapeutic equivalence, a pharmacokinetic equivalence study can be performed. According to the Food and Drug Administration (FDA) guidance for industry,12 pharmacokinetic equivalence or bioequivalence is determined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action, when administered at the same molar dose under similar conditions in an appropriately designed study. In other word, it is the condition where two medicinal products show a similar bioavailability. The objective of the present study (registry identification number: NCT02233777) was to find out whether the pregabalin capsule produced by PT Dexa Medica, Palembang, Indonesia was bioequivalent to the reference product manufactured by Pfizer Manufacturing Deutschland GmbH, Germany.
Methods
Study subjects and design
Before study commencement, the protocol, patient information, and consent form was approved by the Health Research Ethics Committee of the Faculty of Medicine, University of Indonesia. Written informed consent was obtained from each study subject before screening. The conduct of the study conformed to the Declaration of Helsinki,14 Good Clinical Practice,15 and Good Laboratory Practice.16
This was a randomized, open-label, two-period, two-sequence, crossover study under fasting condition, with 1-week washout period, involving 24 subjects. The inclusion criteria were as follows: healthy male or female subject between 18 and 55 years; body mass index ranging from 18 to 25 kg/m2; normal vital signs; and preferably nonsmoker or smoking less than 10 cigarettes per day. Subjects were excluded if they were pregnant or lactating women; had a known contraindication or hypersensitivity to pregabalin; had any liver or renal dysfunction; positive test results for hepatitis B surface antigen (HBsAg), anti-hepatitis C virus (HCV), and/or anti–human immunodeficiency virus (anti-HIV); electrocardiography (ECG) abnormalities; any chronic medical illnesses; any gastrointestinal disease; history of anaphylaxis; history of drug or alcohol abuse within 12 months before screening; any bleeding, coagulation, or other clinically significant hematology disorders; any neurological disorders; intake of any prescription or nonprescription drug, food supplements, or herbal medicines within 14 days of the study’s first dosing day; and participation in any clinical trial within the past 90 days.
Subjects underwent all examinations for screening 14 days before their first dosing day. A pregnancy test, for female subjects, was performed at screening and before taking the drug in each period.
Study products
The test formulation, Leptica® (PT Dexa Medica), and the reference formulation, Lyrica® (Pfizer Inc.), in this study both contain pregabalin 150 mg. In this crossover study, each subject was administered the test formulation and reference formulation according to a random sequence. The randomization code was created using permuted block allocation and a table of random numbers as suggested by Dixon and Massey.17 In this open-label procedure, both subject and investigator knew whether the subject was given the test or reference formulation.
Treatment phase and blood sampling
Subjects were requested to fast except from mineral water, beginning the night before drug administration. Next morning, a 10 mL blood sample from each subject was taken within 1 hour before drug administration. Afterward, subjects were administered the drug, in a sitting posture. The test formulation or reference formulation was given with 200 mL of water and swallowed without chewing. Then, 5 mL postdose blood samples were collected at 20 and 40 minutes, and at 1, 1.5, 2, 2.5, 3, 4, 6, 9, 12, 24, and 36 hours. Blood samples were drawn from subjects’ forearm vein, using a 22 G drawing needle and citrate vacuum tubes or disposable syringe. Then, the samples were collected in polypropylene tubes containing citrate, before centrifugation at 1,538 RCF for 15 minutes to separate the plasma. All plasma samples were transferred to a clean tube and then stored in a freezer at −20°C±5°C until assayed.
During the sampling days, lunch and dinner for subjects were provided 4 hours and 10 hours, respectively, after drug administration. Any xanthine-containing food or beverages, and fruit juices were not allowed to be taken 24 hours before and during the entire sampling days. Subjects had a standard amount of food intake and physical activity during the sampling days. After a 1-week washout period, the same procedure was repeated with the alternate formulation.
Drug concentration analysis
Each plasma sample was dispensed in an appropriate tube, and a gabapentin solution was added as the internal standard before the protein precipitation extraction process.18,19 Afterward, it was extracted with trichloroacetic acid, resulting in a supernatant that was subjected to ultra-performance liquid chromatography with tandem mass spectrometry (UPLC-MS/MS) (Acquity® UPLC H-Class system with Xevo® TQD Detector; Waters Corp, Milford, MA, USA).
The analytical column used in the study was the Acquity UPLC® C18 (1.7 μm, 2.1×50 mm; Waters Corp). The mobile phase was formic acid 0.1% in water and formic acid 0.1% in methanol, with composition arranged in gradient conditions, and the flow rate was set at 0.3 mL/min. A 2 μL sample was automatically injected into the chromatography system by the instrument. The condition of the UPLC-MS/MS system described had been validated with respect to adequate sensitivity, specificity, linearity, accuracy, and precision (both within and between days). The validation data is presented in Table 1.
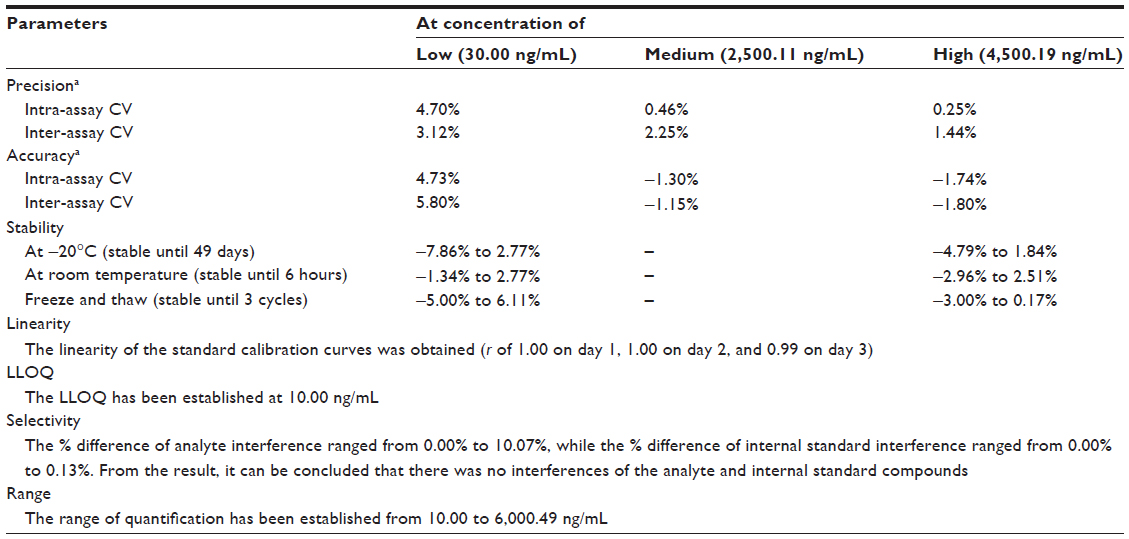 | Table 1 Validation data for the analytical method used for the determination of pregabalin in human plasma by UPLC-MS/MS using gabapentin as the internal standard |
Pharmacokinetic analysis
From the analysis of the plasma concentration time data, several pharmacokinetic parameters were derived. The Cmax and tmax were obtained directly from the observed data. The area under plasma the concentration–time curve from time zero to last observed quantifiable concentration (AUC0–t) was calculated by trapezoidal method. The area under the plasma concentration–time curve from time zero to infinity (AUC0–∞) and t1/2 were calculated according to the formula given in the FDA guidance for industry.12
Statistical analyses for the pharmacokinetic parameters of pregabalin were performed by using analysis of variance (ANOVA) (Excel® 2010; Microsoft Corp, Redmond, WA, USA), after transformation of the data to their logarithmic values. The 90% confidence intervals (CIs) were obtained after back-transformation of the result from the calculation of the logarithmic data values, using the available formula.20
Between-group difference in tmax was analyzed nonparametrically on the original data using the Wilcoxon matched-pairs test, while the t1/2 difference was analyzed using Student’s paired t-test. The power of study was 80%, with an alpha level of 0.05 (two-sided). The acceptance criteria for bioequivalence were 90% CIs of geometric mean ratios between 0.80 and 1.25 for the AUC and Cmax.
Results
Of 24 subjects enrolled in the study, there were four subjects withdrawn during the first period because of vomiting, leaving 20 subjects available for pharmacokinetic analysis. All subjects included in the study were healthy Indonesians who had normal values of vital signs and laboratory parameters. There were 15 males and five females, aged between 19 and 45 years with a body mass index between 18.56 and 24.91 kg/m2, whose blood samples were analyzed for determination of pregabalin concentrations and pharmacokinetic parameters after a single-dose oral administration of the test or reference formulation.
The values of the pharmacokinetic parameters and 90% CIs for geometric mean ratios of the test formulation/reference formulation for pregabalin are presented in Table 2. The mean pregabalin concentrations in 20 subjects after a single-dose oral administration of test or reference formulation are plotted in Figure 2.
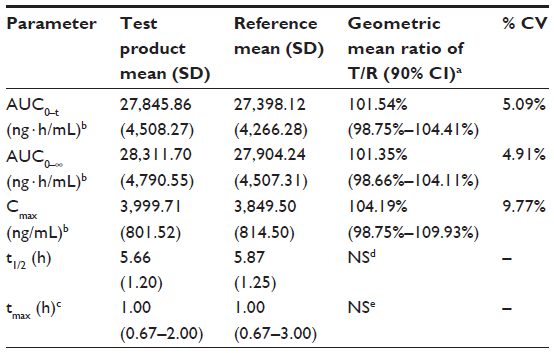 | Table 2 Pharmacokinetic parameters and statistical comparison of pregabalin, after single-dose oral administration of pregabalin capsule 150 mg test or reference formulation |
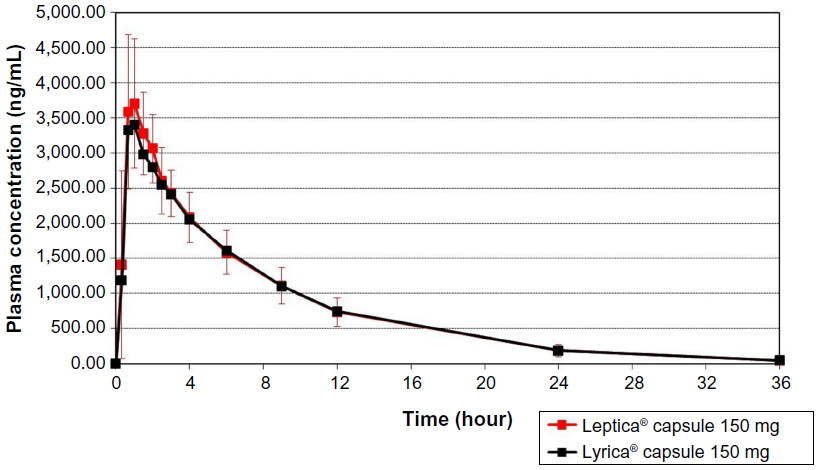 | Figure 2 The mean pregabalin concentrations in 20 subjects, after a single-dose oral administration of the test formulation, Leptica® (PT Dexa Medica, Palembang, Indonesia), or the reference formulation, Lyrica® (Pfizer Manufacturing Deutschland GmbH, Germany). |
Of all enrolled subjects, 13 of the subjects that received test formulation and 15 of the subjects that received reference formulation experienced adverse events, with the most frequently occurring being lightheadedness (16 events), dizziness (12 events), nausea (seven events), vomiting (four events), and drowsiness (two events). All four subjects with a vomiting event in the first period – among whom three had received the reference formulation and one had received the test formulation – withdrew their participation in the study. There was no serious adverse event reported in the study, and all events were resolved at the end of the study.
Discussion
The objective of this study was to evaluate whether the test formulation of pregabalin capsule 150 mg is bioequivalent to the reference formulation. Since absorption of the investigational formulation is sufficient to measure the drug concentration directly in blood and systemic absorption is relevant to drug action, a blood-level bioequivalence study was preferred over a pharmacologic or clinical end point study.21 The results were mainly intended to fulfil regulatory requirements for an abbreviated new drug application (ANDA) submission of the tested product. The condition of bioequivalence demonstrates that the generic medicinal product is therapeutically equivalent with its reference, and therefore, it is unnecessary to conduct any comparative clinical studies to prove safety and efficacy. Thus, selection of the reference product is important. Reference products should pass through a series of preclinical and clinical trials in order to establish their safety and efficacy.22,23 Because the outcome from one bioequivalence study cannot be generalized to any other generic medicinal products containing the same active substance, the present study was required despite the availability of bioequivalence studies on other pregabalin capsule formulations.19,24
A further intention in performing a reliable bioequivalence study is to provide a generic product that, not only offers more competitive pricing but also, demonstrates similar quality to its reference formulation in regard to safety and efficacy. The concept of providing a generic medicinal product supports the application of pharmacoeconomics. According to Arenas-Guzman et al,25 pharmacoeconomics is the branch of economics related to the most economical and efficient use of pharmaceuticals to yield maximum value to patients, health care payers, and society in general. Recently, health care practitioners have been encouraged to make clinical and policy decisions based on the consideration of economic aspects rather than on clinical outcomes only. One of the most fundamental assessments is the assessment of direct medical costs, one of which is drug cost.26 The availability of a generic medicinal product can give pharmacoeconomic benefits according to patient and provider perspectives because it gives option for both patient and the health care provider when they have to select the most rational medication.
This bioequivalence study was conducted in healthy subjects in order to minimize pharmacokinetic variability, which can emerge if performed in patients with any concomitant illnesses and medications. The number of subjects that participated in the study was determined using means of CIs as tabulated by Diletti et al.27 The intrasubject variability, reported as CV, obtained for the pregabalin AUC0–t was 5.09%. With this result, the participation of 20 subjects in the study was regarded adequate to ensure that the study had enough power to confirm statistical conclusion.
In this study, the study drugs were given as a single-dose preparation as recommended in the guidance of bioavailability and bioequivalence studies12 because this is generally more sensitive in assessing release of drug substance into the systemic circulation. For a single-dose study, measurement of the study drug’s total exposure will include AUC0–t and AUC0–∞; while measurement of peak exposure will comprise the Cmax, which is obtained directly from the systemic drug concentration data. The results of the present study showed that the 90% CIs of the test/reference ratios for AUC0–t, AUC0–∞, and Cmax of pregabalin were within the acceptance range of bioequivalence (80.00%–125.00%).
Besides AUC and Cmax, this study also reported the tmax and t1/2 of pregabalin. The median (range) of tmax for the test and reference formulations of pregabalin were 1.00 (0.67–2.00) hours and 1.00 (0.67–3.00) hours, respectively. Those values were proven not significant statistically. In the meantime, the mean (SD) of the t1/2 for the test and reference formulations of pregabalin were 5.66 (1.20) hours and 5.87 (1.25) hours, respectively. These values were slightly lower than those found in literature, but they were not significantly different, demonstrating a comparable rate of drug elimination from the body, between the test and reference formulations.
Conclusion
The present study concluded that the two formulations of pregabalin capsule 150 mg are bioequivalent.
Acknowledgments
The authors deeply thank all volunteers for their participation in this study. The assistance of the clinical investigator Danang Agung Yunaidi, who provided and cared for the study subjects, is gratefully appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
Sweetman SC, editor. Martindale: The Complete Drug Reference. 36th ed. London: Pharmaceutical Press (PhP); 2009. | |
American Society of Health-System Pharmacists, McEvoy GK, Snow EK, eds. AHFS Drug Information 2013. Bethesda, Maryland: Board of the American Society of Health-System Pharmacists; 2013. | |
Finnerup NB, Jensen TS. Clinical use of pregabalin in the management of central neuropathic pain. Neuropsychiatr Dis Treat. 2007;3(6):885–891. | |
Blommel ML, Blommel AL. Pregabalin: an antiepileptic agent useful for neuropathic pain. Am J Health Syst Pharm. 2007;64(14):1475–1482. | |
Bockbrader HN, Burger P, Knapp L, Corrigan BW. Population pharmacokinetics of pregabalin in healthy subjects and patients with chronic pain or partial seizures. Epilepsia. 2011;52(2):248–257. | |
Ben-Menachem E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia. 2004;45(6):13–18. | |
who.int [homepage on the Internet]. Epilepsy. Fact sheet number 999. World Health Organization; 2015 [cited September 12, 2014]. Available from: http://www.who.int/mediacentre/factsheets/fs999/en/. Accessed March 11, 2015. | |
Commission on Tropical Diseases of the International League against Epilepsy. Relationship between epilepsy and tropical diseases. Epilepsia. 1994;35(1):89–93. | |
Burneo JG, Tellez-Zenteno J, Wiebe S. Understanding the burden of epilepsy in Latin America: a systematic review of its prevalence and incidence. Epilepsy Res. 2005;66(1–3):63–74. | |
Preux PM, Druet-Cabanac M. Epidemiology and aetiology of epilepsy in sub-Saharan Africa. Lancet Neurol. 2005;4(1):21–31. | |
Mac TL, Tran DS, Quet F, Odermatt P, Preux PM, Tan CT. Epidemiology, aetiology, and clinical management of epilepsy in Asia: a systematic review. Lancet Neurol. 2007;6(6):533–543. | |
Center for Drug Evaluation and Research. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. Washington, DC: Food and Drug Administration, US Department of Health and Human Services; 2003. Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm070124.pdf. Accessed September 12, 2014. | |
Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. London: European Medicines Agency; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed September 12, 2014. | |
wma.net [homepage on the Internet]. World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. World Medical Association; 2013 [cited September 12, 2014]. Available from: http://www.wma.net/en/30publications/10policies/b3/. Accessed March 11, 2015. | |
ICH Expert Working Group. Guideline for Good Clinical Practice – ICH Harmonised Tripartite Guideline. Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 1996. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed September 12, 2014. | |
Chemicals Group and Management Committee, Environment Directorate. OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring, Number 1 – OECD Principles on Good Laboratory Practice. Paris: Organisation for Economic Co-operation and Development; 1998. Available from: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/mc/chem(98)17&doclanguage=en. Accessed September 12, 2014. | |
Dixon WJ, Massey FJ. Introduction to Statistical Analysis. 3rd ed. New York, NY: McGraw-Hill; 1969. | |
Mandal U, Sarkar AK, Gowda KV, et al. Determination of pregabalin in human plasma using LC-MS/MS. Chromatographia. 2008;67(3–4):237–243. | |
Quiñones L, Sasso J, Tamayo E, et al. A comparative bioavailability study of two formulations of pregabalin in healthy Chilean volunteers. Ther Adv Chronic Dis. 2010;1(4):141–148. | |
Badan Pengawas Obat dan Makanan Republik Indonesia (BPOM RI). Pedoman uji bioekivalensi [Guideline for bioequivalence studies]. Jakarta: BPOM RI; 2004. Bahasa. Available from: http:// jdih.pom.go.id/produk/PERATURANKEPALABPOM/PER KBPOM_NO.HK.00.05.3.1818 TH 2005_Tentang PEDOMANUJI BIOEKIV_2005.pdf. Accessed March 30, 2015. | |
Kesselheim AS, Misono AS, Lee JL, et al. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. JAMA. 2008;300(21):2514–2526. | |
Thiessen JJ. Chapter 8: Bioavailability and bioequivalence. In: du Souich P, Orme M, Erill S, editors. The IUPHAR Compendium of Basic Principles for Pharmacological Research in Humans. Kansas City, KS: International Union of Pharmacology (IUPHAR ); 2004:55–66. Available from: http://www.iuphar.org/pdf/hum_55.pdf. Accessed September 12, 2014. | |
Jang KH, Seo JH, Yim SV, Lee KT. Rapid and simple method for the determination of pregabalin in human plasma using liquid chromatography-tandem mass spectrometry (LC-MS/MS): application to a bioequivalence study of Daewoong pregabalin capsule to Lyrica® capsule (pregabalin 150 mg). J Pharm Invest. 2011;41(4):255–262. | |
Center for Veterinary Medicine. Guidance for industry: Bioequivalence Guidance. Washington, DC: Food and Drug Administration, US Department of Health and Human Services; 2006. Available from: http://www.fda.gov/downloads/AnimalVeterinary/Guidance ComplianceEnforcement/GuidanceforIndustry/ucm052363.pdf. Accessed September 12, 2014. | |
Arenas-Guzman R, Tosti A, Hay R, Haneke E; National Institute for Clinical Excellence. Pharmacoeconomics – an aid to better decision-making. J Eur Acad Dermatol Venereol. 2005;19 Suppl 1:S34–S39. | |
Trask LS. Pharmacoeconomics: principles, methods, and applications. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey M, editors. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York: McGraw-Hill Global Education Holdings. Available from: http://accesspharmacy.mhmedical.com/content.aspx?bookid=462§ionid=41100767. Accessed February 26, 2015. | |
Diletti E, Hauschke D, Steinijans VW. Sample size determination for bioequivalence assessment by means of confidence intervals. Int J Clin Pharmacol Ther Toxicol. 1991;29(1):1–8. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.