")
Back to Archived Journals » Clinical Oncology in Adolescents and Young Adults » Volume 4
Ewing sarcoma in adolescents and young adults: diagnosis and treatment
Authors Davis L, Malempati S
Received 28 January 2014
Accepted for publication 16 April 2014
Published 1 September 2014 Volume 2014:4 Pages 21—31
DOI https://doi.org/10.2147/COAYA.S61451
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Lara E Davis,1,2 Suman Malempati2
1Department of Medicine, 2Department of Pediatrics, Oregon Health and Science University, Portland, OR, USA
Abstract: Ewing sarcoma (ES) is a rare tumor that most often occurs in adolescents and young adults. This review discusses the diagnosis, prognosis, and treatment of localized and metastatic ES, with an emphasis on the care of adolescent and young adult patients. The pathobiology is reviewed. Particular attention is given to recent and current clinical trials, and an introduction to future directions for therapy of ES is provided.
Keywords: Ewing, sarcoma, primitive neuroectodermal tumor, PNET
Introduction
The Ewing sarcoma family of tumors (ESFTs) includes small, round, blue cell tumors with a characteristic translocation involving EWS and an ETS-family gene, most frequently FLI1. These tumors include ES of bone, extraskeletal ES, peripheral primitive neuroectodermal tumors (PNET), and Askin’s tumors (PNET of the chest wall).
Epidemiology
Approximately 50% of ESFTs occur in adolescents and young adults (AYAs).1 ES is uncommon, however, and thus accounts for less than 5% of the malignancies diagnosed in AYA patients.1 The peak incidence of ES occurs between 10 and 20 years of age, and ES is rare over the age of 30 years.2
Among persons 15–29 years old, there are approximately 260 ESs diagnosed each year in the US.1 Surveillance, Epidemiology, and End Results data indicate that within adolescents 15–19 years old, males are nearly twice as likely to be diagnosed with ES than females, and Caucasian teens develop ES 18 times more frequently than African Americans (Figure 1).3 These statistics are consistent worldwide, including the rarity of ES in black populations.4 The reason for the relatively low incidence of ES in black populations is unknown.
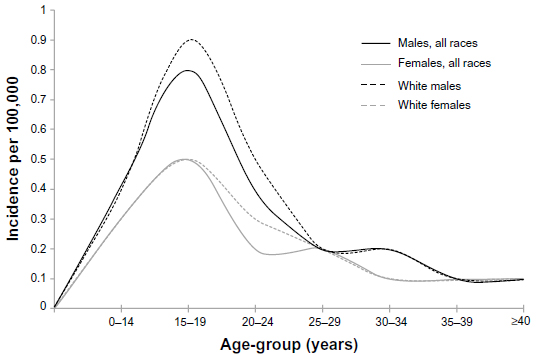 | Figure 1 Incidence of Ewing sarcoma within the adolescent and young adult population, 2004–2008. |
Clinical presentation
ES typically presents as a rapidly growing mass that is often painful. The long bones are the most common primary sites, although the bones of the axial skeleton are also frequently involved, particularly in older patients. ES can arise anywhere in the body, and up to 20% of cases are extraskeletal.2
Typical radiographic characteristics of aggressive bone malignancies are seen in ES, including a permeative pattern of bony destruction, often through the marrow space and/or with an associated soft-tissue mass. The classic appearance of lamellate or “onion skin” periosteal reaction is frequently seen on radiography, and represents the layering of new bone surrounding a region of malignant cortical destruction (Figure 2).
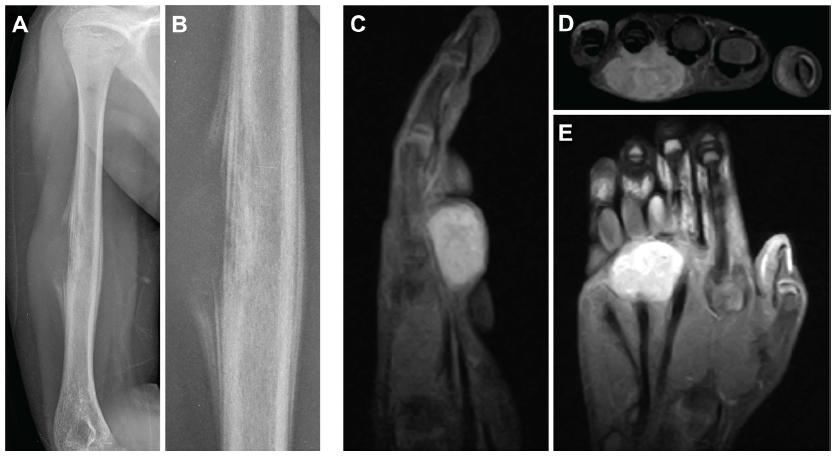 | Figure 2 (A–E) Radiologic appearance of Ewing sarcomas. |
Staging
Approximately 20%–25% of patients will have metastatic disease detected at initial diagnosis.5–7 Of those patients with primary metastatic disease, an equal percentage present with metastases limited to the lungs versus limited to bones and/or bone marrow (approximately 40% each), while an additional 20% have disease in multiple distant sites.7 Therefore, at a minimum, initial staging workup should include radiographs and magnetic resonance imaging of the primary site, as well as computed tomography of the chest and a whole-body bone scan. 18F-fluorodeoxyglucose positron-emission tomography may be considered, but is not universally recommended. Staging of ES follows the TNM (tumor, nodes, metastasis) system for bone tumors.8 However, apart from metastatic versus localized disease, the disease stage is not clinically relevant for prognosis or treatment.
Diagnosis
The diagnoses of typical ES, extraskeletal ES, and PNET were not widely recognized to be a single pathologic entity until the early 2000s, at which point genomic data supported the grouping of all EWS-ETS-driven tumors together.
Histology and immunohistochemistry
The diagnosis of ES can often be made by core needle biopsy, although open biopsy may be necessary in atypical cases. ESFTs are high-grade tumors by definition.2 Historically, the term PNET was used for tumors with histologic features of neuroectodermal differentiation, while the term ES was applied to tumors lacking such differentiation.1,2 Typical ES has a homogeneous appearance with sheets of small round blue cells, and may contain variable amounts of necrosis (Figure 3). PNETs may have the additional finding of rosettes or pseudorosettes, indicative of neural differentiation.2 Atypical ES includes histologic variants, such as those with significant cellular and nuclear pleomorphism or hemangioendothelial features.9
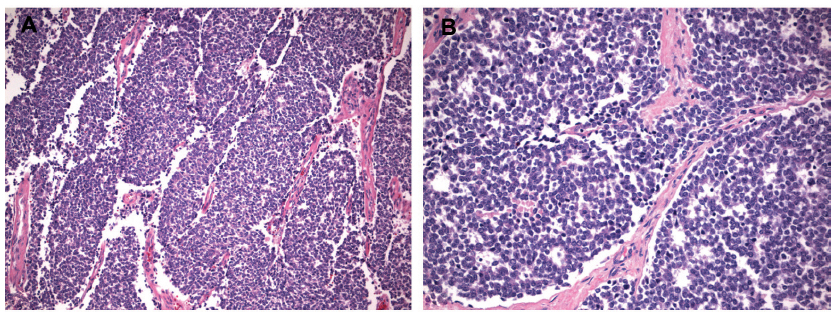 | Figure 3 Hematoxylin and eosin stain of typical Ewing sarcoma/primitive neuroectodermal tumor at 10× (A) and 20× (B) magnification. |
The vast majority of ESFTs are CD99-positive, predominantly in a membranous pattern, but cytoplasmic positivity may also be present. In cases where additional immunohistochemistry confirmation is needed, positive nuclear Fli1 or cytoplasmic and nuclear caveolin 1 expression may be helpful. However, these are rarely used in practice, since disruption of EWSR1 can be readily identified by fluorescence in situ hybridization.9
Translocation
The ESFTs are defined by a characteristic translocation involving the central exons of the EWSR1 gene on chromosome 22 and the central exons of an ETS-family gene. There are 29 members of the ETS (E26 transformation-specific) family of transcription factors, which are proto-oncogenes as a result of their important roles in cell-cycle control. A tumorigenic fusion protein forms when one of these transcription factors is aberrantly under the control of a transcription activation domain from within the EWS gene.
Approximately 85% of ESs result from t(11;22), which produces the EWSR1-FLI1 oncoprotein.2 While translocations with FLI1 are most common, an additional 10% of ESFTs are associated with a translocation with ERG on chromosome 21.10,11 Alternative proto-oncogene fusion partners include ETV1, ETV4 (E1AF), and FEV, among others.12–14 The specific translocation or fusion type is no longer thought to have prognostic significance.15
By itself, lack of molecular confirmation of EWSR1 rearrangement does not rule out a diagnosis of ES. Rearrangements of other TET-family genes may also result in ES, and there is a growing body of literature on “Ewing-like” sarcomas that harbor translocations between TET-family genes and non-ETS genes, or non-TET genes and ETS-family genes.2 A summary of chromosomal rearrangements found in ES and Ewing-like sarcoma is shown in Table 1.
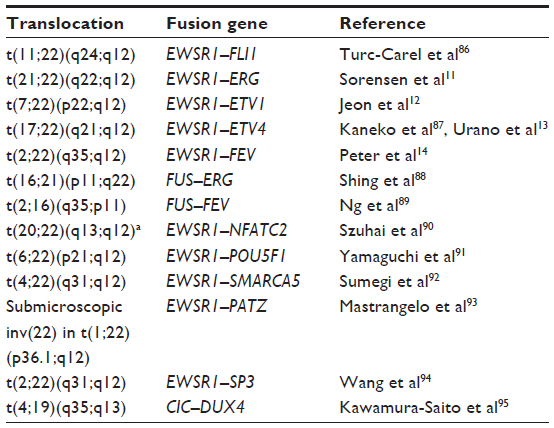 | Table 1 Chromosomal rearrangements found in Ewing sarcoma and “Ewing-like” sarcoma |
Prognosis
The presence or absence of metastatic disease is the single most powerful predictor of outcome in ES.5 Event-free survival (EFS) at 5 years is greater than 70% for patients with localized disease, but has stagnated at 30% for patients with primary metastatic disease.7,16–18 Among those with metastases, better prognosis is associated with metastases confined to the lung; patients with unilateral pulmonary metastases do better still.6,7,19,20
While risk stratification of ES has not entered into routine treatment decisions, high-risk localized disease is generally defined as any pelvic tumor, tumors that cannot achieve complete local control (details in the “Treatment” section), or large tumors with poor histologic response to preoperative therapy.
The primary tumor site and size are well-recognized prognostic factors. In general, more distal tumors predict better outcomes compared to tumors located more centrally. Pelvic and sacral ES have the worst outcomes, with 5-year EFS approximately 50%.6,16,17,19 Larger tumors, generally defined as those ≥8 cm (or 200 mL in volume), portend a similar reduction in EFS.19,21
Response to preoperative chemotherapy is also strongly predictive of disease-free survival. Patients with <5% viable tumor at the time of resection are dramatically more likely to be disease-free after 5 years compared to patients with ≥30% viable tumor (disease-free survival 75% versus 20%, P<0.001).22
In addition, older age is a negative prognostic indicator. Young children have the best outcomes, while adults over the age of 40 years fare the worst. A summary of outcomes by age reported in both prospective and retrospective studies is provided in Table 2.
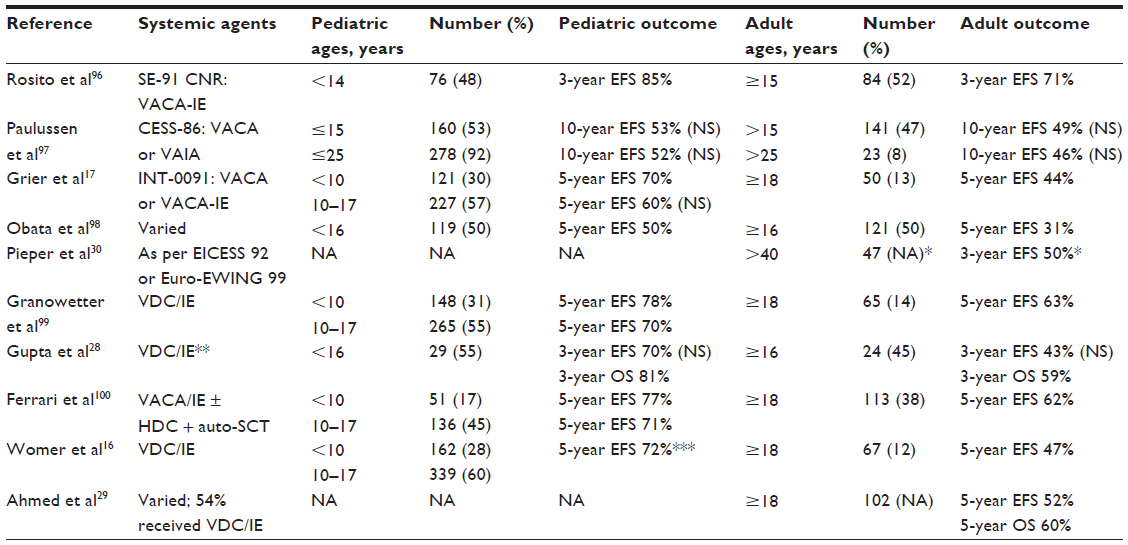 | Table 2 Outcomes by age for localized Ewing sarcoma |
Finally, patients with recurrent disease have a dismal prognosis. Of the 30%–40% of patients with disease relapse after initial treatment, less than 20% will survive 5 years following recurrence.23 Longer survival may be seen in relapsed patients with favorable features, such as limited local recurrence or recurrence occurring more than 2 years after initial diagnosis.23
Treatment
Patients with ESFTs should be evaluated and treated at a sarcoma center with multidisciplinary expertise. The importance of diagnosis and treatment by a multidisciplinary team is noted within the National Comprehensive Cancer Network Guidelines, among others.24–27
Localized disease
The current standard of care in North America for localized ES was established by large cooperative clinical trials completed in the 1990s and early 2000s. In the landmark Intergroup (INT)-0091 trial, the addition of ifosfamide and etoposide (IE) to a backbone of vincristine, doxorubicin, and cyclophosphamide (VDC) improved overall survival (OS) among patients without metastases from 61% to 72% (P=0.01), and EFS improved from 54% to 69% (P=0.005, Figure 4).17 The standard of care was further refined by alternating IE with VDC every 2 weeks rather than every 3 weeks (termed interval compression or dose-dense) following completion of the Children’s Oncology Group (COG) trial AEWS0031.16 In this trial, interval compression improved 5-year EFS from 65% to 73% (P=0.05), and showed a trend toward improvement in OS (77% versus 83%, P=0.06, Figure 5A and B).
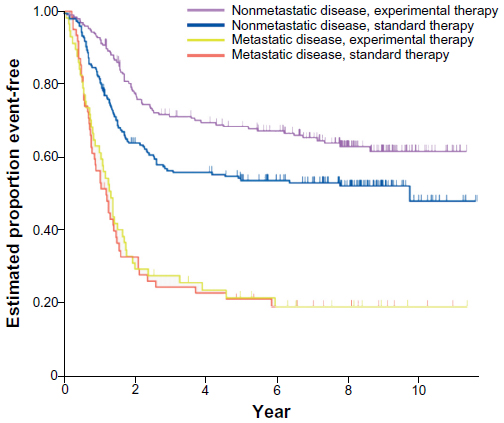 | Figure 4 Results of the Intergroup (INT)-0091 trial of four-drug versus six-drug chemotherapy regimens: event-free survival according to study group and the presence or absence of metastatic disease. |
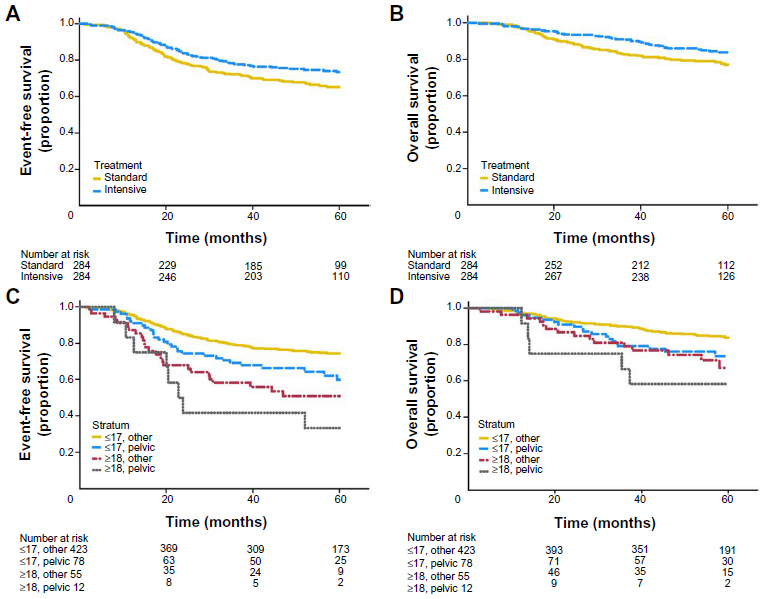 | Figure 5 (A–D) Results of the Children’s Oncology Group AEWS0031 trial of interval compression. |
The majority of patients in these trials were pediatric, but patients over the age of 18 years were eligible. Patients 18 years or older enrolled in AEWS0031 (n=67) experienced a considerably worse EFS of 47% versus 72% at 5 years (P<0.001, Figure 5C).16 Poorer outcomes for patients with pelvic primaries were redemonstrated in this trial as well, such that pediatric patients with pelvic tumors had equivalent outcomes to adult patients with nonpelvic primaries. A single-institution retrospective analysis of patients with localized, nonpelvic ES treated within the adult cancer center (generally patients >16 years old) compared to children treated within the affiliated pediatric center, found a significant difference in 3-year OS of 67% versus 88% (P=0.04).28 However, similar analyses at a different institution found 5-year OS and EFS among adult patients to be comparable to those achieved in INT-0091, and a retrospective subset analysis from two modern European cooperative group trials suggests that adults over the age of 40 years can have similar outcomes to adolescents when treated in strict accordance with a pediatric protocol.19,29,30 Taken together, these data suggest that with contemporary supportive care, adults with localized, nonpelvic primary tumors may achieve outcomes similar to children if treated with interval-compressed multidrug regimens and aggressive local control measures (Table 2).
Local control with surgery and/or radiation is paramount. Whether surgery achieves better local control than radiation therapy alone is controversial, but in practice, surgery is generally preferred when possible due to the known late effects of radiation.31–33 Local control is generally undertaken after 12 weeks of “induction” chemotherapy with VDC/IE.16 For residual disease following surgical resection, postoperative radiation to 45–50.4 Gy is recommended, and can be administered concurrently with IE, preferably beginning within 2 weeks (and not more than 6 weeks) of resection. ESs are very radiosensitive, and radiation should always be employed when complete surgical resection is not feasible. In these cases, definitive radiation therapy to a dose of 45 Gy to the tumor bed is recommended, with an additional 10.8 Gy to a surrounding margin of 1.5 cm minimum.16
Metastatic disease
There is no broadly accepted standard of care for metastatic ES, and numerous treatment regimens are actively being investigated (see the “Future directions” section). All patients with metastatic ES should be offered enrollment in a clinical trial.
Nonetheless, there is evidence that the intensification approaches taken for localized disease do not benefit those with metastases. The INT-0091 trial demonstrated that there is no benefit to the addition of IE to the standard three-drug regimen of VDC for patients with metastatic disease (Figure 4).17 There is also no evidence that interval compression improves outcomes, except in patients with localized disease.16 Therefore, outside a clinical trial, a frontline regimen of VDC every 3 weeks is reasonable for these patients.
Various multichemotherapy regimens for relapsed and refractory ES are well represented in the literature in retrospective institutional experiences or small clinical trials. Irinotecan and temozolomide (with or without vincristine) or cyclophosphamide and topotecan administered for 5 days every 3 weeks are reasonable outpatient regimens with published efficacy and tolerability.34–37 Treatment with numerous additional systemic agents and regimens have been reported, including the synthetic alkaloid PM00104, VEGFR inhibitors, or gemcitabine/docetaxel, but none has demonstrated sufficient improved efficacy or reduced toxicity sufficient to supplant the current standards of care.38–40
For patients with lung metastases, whole-lung irradiation is sometimes employed, although prospective data are lacking.7,20,41 There are data to support aggressive local treatment of metastatic sites (surgical resection or radiotherapy) when possible.42,43 High-dose chemotherapy followed by autologous stem cell transplantation (SCT) has been investigated repeatedly, but remains controversial. The results of numerous studies are mixed, and there is no consensus that this therapy provides benefit, given the biases inherent in the studies published to date.44–46 There is therefore insufficient evidence currently to support treatment with autologous transplant outside a clinical trial. Allogeneic transplant has been used rarely, although there are isolated reports of long-term survivors of metastatic ES using this approach.47–49 The Euro-EWING (Ewing tumour Working Initiative of National Groups)-99 trial (described in the “Current clinical trials” section) attempts to resolve the debates over whole lung irradiation and autologous SCT in metastatic ES definitively, but results are not yet available.
Surveillance
Upon completion of curative-intent chemotherapy, the COG recommends radiographs of the primary site, plus chest computed tomography and a whole-body bone scan. Follow-up radiographs of chest and the primary site are recommended for the subsequent 10 years, initially every 3 months, then decreasing in frequency steadily to annually for the final 5 years.50 ES has the potential to recur several years after initial remission.17,51 Therefore, prolonged surveillance for recurrent disease is necessary.
Survivors of ES require ongoing long-term follow-up care to monitor for late effects of therapy. Historically, patients with ES were among the most likely to develop a treatment-related second malignancy, often sarcomas.52 A large percentage of these cancers were likely radiation-associated, and as local treatment has shifted towards surgery, the long-term effects of systemic therapy are becoming more prominent. A patient treated with the standard interval-compressed five-drug regimen for localized ES will have received cumulative doses totaling doxorubicin 375 mg/m2, cyclophosphamide 10.8 g/m2, ifosfamide 72 g/m2, etoposide 4,000 mg/m2, and 18 doses of vincristine. Secondary myeloid dysplasia/neoplasia is a concern, as are early coronary artery disease, infertility, renal dysfunction, and neuropathy. Cumulative mortality among ES survivors is 25% at 25 years after diagnosis, and survivors have a markedly increased risk of severe, life-threatening, or disabling health conditions (relative risk 6.0) compared to siblings.53
Future directions
Refinements to interval-compressed five-drug chemotherapy are expected, but novel treatment approaches targeting the pathogenic molecular pathways underlying ES will be moving into the clinic in the foreseeable future as well. In order for these advances to positively impact outcomes for AYAs, it is imperative that ES patients of all ages are enrolled in clinical trials and biospecimen banking. There has been a push in recent years to open clinical trial enrollment for “pediatric” diseases to AYA-aged patients. As a result, AYAs (including up to and sometimes beyond age 40 years) are eligible for the majority of current ES clinical trials and biology studies.
Current clinical trials
The COG and Spain’s Grupo Español de Investigación en Sarcomas (GEIS) have large, multi-institutional ES trials ongoing, investigating alternative cytotoxic regimens for localized disease (NCT01231906, NCT01696669). The GEIS study incorporates gemcitabine and docetaxel for high-risk patients, defined as patients with primary tumors of the axial skeleton. The COG AEWS1031 study is comparing standard interval-compressed VDC/IE with a similar regimen incorporating topotecan, based on the efficacy of topotecan in the relapsed and metastatic setting.35,54
The poor clinical outcomes for patients with metastatic disease have stimulated numerous investigations into new ways to use old treatments, as well as novel treatments. The current largest effort is directed at high-dose chemotherapy with autologous SCT. This strategy has been investigated in small series for decades, but the large prospective Euro-EWING-99 trial (NCT00020566) is incorporating SCT for high-risk disease. Euro-EWING-99 is a complex international trial that enrolled patients into four separate, risk-stratified cohorts: nonmetastatic with good histologic response to induction chemotherapy, nonmetastatic with poor histologic response, isolated pulmonary metastases, and metastatic disease to sites other than the lungs. Patients with nonmetastatic disease but poor histologic response after six cycles of vincristine, ifosfamide, doxorubicin, and etoposide were randomized to vincristine, actinomycin, and ifosfamide consolidation chemotherapy or high-dose therapy with busulfan and melphalan with autologous peripheral blood stem cell rescue. The COG participated in this trial, but limited enrollment to patients with pulmonary metastases (under protocol AEWS0031); this cohort randomizes patients to vincristine–doxorubicin–ifosfamide consolidation with whole-lung irradiation or high-dose therapy with busulfan–melphalan and autologous SCT. This study will likely close soon, and results are not yet published.
The morbidity of these intensive chemotherapy-treatment regimens is significant, including therapy-related deaths as well as long-term sequelae. There is thus parallel interest in minimizing toxicity of treatment. Small-molecule-targeted therapies hold promise for improving outcomes with minimal additional toxicity.
The importance of insulin-like growth factor (IGF) signaling in ES has been repeatedly demonstrated in molecular biology investigations, and thus there was significant excitement among ES clinicians and researchers upon the development of clinical IGF 1 receptor (IGF1R) inhibitors.44,55–59 A subset of patients appears to benefit from IGF1R inhibition, but among the general ES population, efficacy is unimpressive. A phase II Sarcoma Alliance for Research through Collaboration (SARC) trial of R1507 (an anti-IGF1R monoclonal antibody that is no longer in clinical development) reported an underwhelming 10% overall response rate.60 Twenty-two adult patients (age range 20–77 years) were enrolled in a Phase II trial of the monoclonal anti-IGF1R antibody ganitumab (AMG479), which demonstrated a median progression-free survival of 7.9 months (95% confidence interval 5.4–25.6 months).61 Over 40% experienced partial response or stabilization of disease, and two patients had prolonged benefit (partial response or stable disease ≥24 weeks). More than 100 patients ages 10–62 years were enrolled in a similar trial to assess the efficacy of the IGF1R antibody figitumumab, but only 14% of patients experienced an objective response.62 A COG Phase I study of single-agent cixutumumab, also a monoclonal anti-IGF1R antibody, included a Phase II expansion cohort for refractory ES (NCT00609141, COG ADVL0712 and NCT00831844, COG ADVL0821). This study had disappointing results for the ES subset, with a progression-free survival of only 44 days.63 Surprisingly, there was also no apparent correlation between tumor expression of IGF1, IGF2, or IGF1R and response to therapy.
A follow-up Phase II combination study of cixutumumab and the mammalian target of rapamycin (mTOR) inhibitor temsirolimus for refractory ES and other sarcomas has not yet reported results (NCT01614795, COG ADVL1221). Preclinical data from the National Cancer Institute (NCI)’s Pediatric Preclinical Testing Program predicts efficacy for this combination in both ES and osteosarcoma, while data from other laboratories questions the utility of targeting IGF1R in ES altogether.64,65 COG will open AEWS1221 in the coming months to define the feasibility of adding ganitumab to interval-compressed VDC/IE for patients with newly diagnosed metastatic ES.66 While the overall response rate to IGF1R inhibitors is disappointing, a few patients have had dramatic responses. Biomarker analysis has been challenging, and has not provided conclusive evidence regarding which patients are likely to respond to IGF1R inhibition. Further biomarker analysis is planned in AEWS1221.
Interest in mTOR has been gathering for years, following the recognition that mTOR signaling plays an important role in ES pathogenesis. Treatment of ES cell lines with rapamycin, the prototypical mTOR inhibitor, results in cell-cycle arrest at G1 and downregulation of EWS–FLI1.67 The aforementioned Phase II study of concurrent IGF1R and mTOR inhibition was based on preclinical data supporting this combination approach. Pediatric Preclinical Testing Program studies of rapamycin demonstrated only intermediate efficacy against ES xenografts when used alone, but were more promising when used together with cixutumumab or vincristine.64,68,69 Seven patients with refractory ES were enrolled in COG ADVL0918, a Phase II trial of temsirolimus in combination with irinotecan and temozolomide.70 One patient sustained an objective response for more than 10 months, while another experienced stable disease for several months.
Poly(adenosine diphosphate ribose) polymerase (PARP)-1 is the small-molecule target drawing the most attention currently in the preclinical realm. PARP-1 is an enzyme important for base-excision repair as well as a key ETS protein-family cofactor, a mechanism first discovered in ETS fusion-positive prostate carcinomas.71 Inhibition of PARP-1 results in decreased anchorage-independent growth and invasion (by in vitro soft-agar colony formation and Boyden transwell experiments) in cell lines expressing EWS–ETS fusions.72 In mouse xenograft models, the PARP-1 inhibitor olaparib slowed tumor growth when given as monotherapy, but when given in combination with temozolomide resulted in immediate tumor regression and sustained complete response, which is nearly unheard of in models of this disease.72 These dramatic results were not replicated in a similar experiment from another laboratory, although response was still seen.73 This combination approach is currently being investigated in a Phase I trial (NCT01858168, the upcoming SARC025), and the COG will likely incorporate a PARP inhibitor into the next Phase II trial for relapsed ES.
Future treatments
Despite the widely held belief that such transcription-factor fusion-protein targets as EWS–FLI1 are “undruggable”, additional promising therapies are on the horizon. High-throughput screening techniques have identified two in particular: mithramycin and YK-4-279.
Mithromycin is a nonspecific antineoplastic antibiotic that cross-links guanine–cytosine-rich chromatin regions and prevents hypermethylation, resulting in altered gene transcription.74–78 It was used clinically in the 1960s–1970s, including in some cases of ES, and has recently been resurrected.79,80 A group of researchers at the NCI reported impressive inhibition of EWS–FLI1 downstream targets and significant in vivo (xenograft) tumor response with mithramycin, and the NCI subsequently opened a Phase II trial of mithramycin monotherapy in EWS–FLI1-positive ES (NCT01610570).81
While the pathognomonic fusion protein in ES was characterized decades ago, no drug specific to this target has successfully been developed. Erkizan et al have identified the most promising candidate to date, a compound that blocks the binding of ribonucleic acid helicase A to the EWS–FLI1 protein and therefore interferes with its oncogenic function.82 YK-4-279 is an optimized derivative of a molecule identified through a surface plasmon-resonance screen of 3,000 small molecules in the NCI Drug Targeting Program library. This compound demonstrates single enantiomer efficacy and minimal toxicity in preclinical animal models, both facts that support the eventual advancement of YK-4-279 into clinical trials.83 Subsequently, this group used a similar process combined with phage display to identify the peptide ESAP1, which potently binds to and inhibits EWS–FLI1.84 Therapeutic peptides, such as ESAP1, are not yet optimized for clinical use due to stability issues, but will likely be in clinical trials within the next decade.
Conclusion
ES is a rare tumor that occurs most frequently in AYAs, and is characterized by disruption of the EWSR1 gene. Diagnosis and treatment require the involvement of multidisciplinary teams at specialized centers. Outcomes for localized disease are good, with 5-year EFS >70% for children. There is evidence that AYAs have similar outcomes when treated according to pediatric regimens. Nonetheless, long-term survivors have significant treatment-related morbidity. Current clinical trials aim to define the optimal treatment for metastatic disease, while preclinical research and early phase trials are investigating several promising new targeted agents.
Acknowledgments
The authors wish to thank Drs Atiya Mansoor, Phil Raess, and Don Sauser for providing clinical images.
Disclosure
The authors report no conflicts of interest in this work.
References
Herzog CE. Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol. 2005;27(4):215–218. | |
International Agency for Research on Cancer. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Geneva: WHO; 2013. | |
US Department of Health and Human Services. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program, 1975–1995. Bethesda (MD): National Cancer Institute; 1999. | |
Parkin DM, Stiller CA, Nectoux J. International variations in the incidence of childhood bone tumours. Int J Cancer. 1993;53(3):371–376. | |
National Cancer Institute. Ewing sarcoma treatment (PDQ®). 2013. Available from: http://www.cancer.gov/cancertopics/pdq/treatment/ewings/HealthProfessional. Accessed January 17, 2014. | |
Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–3114. | |
Paulussen M, Ahrens S, Burdach S, et al. Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol. 1998;9(3):275–281. | |
Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti, editors. AJCC Cancer Staging Manual. 7th ed. New York: Springer; 2010. | |
Llombart-Bosch A, Machado I, Navarro S, et al. Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009;455(5):397–411. | |
Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin Cancer Res. 2010;16(16):4077–4083. | |
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6(2):146–151. | |
Jeon IS, Davis JN, Braun BS, et al. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene. 1995;10(6):1229–1234. | |
Urano F, Umezawa A, Hong W, Kikuchi H, Hata J. A novel chimera gene between EWS and E1A-F, encoding the adenovirus E1A enhancer-binding protein, in extraosseous Ewing’s sarcoma. Biochem Biophys Res Commun. 1996;219(2):608–612. | |
Peter M, Couturier J, Pacquement H, et al. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene. 1997;14(10):1159–1164. | |
Le Deley MC, Delattre O, Schaefer KL, et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-EWING 99 trial. J Clin Oncol. 2010;28(12):1982–1988. | |
Womer RB, West DC, Krailo MD, et al. Randomized Controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(33):4148–4154. | |
Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. | |
Cangir A, Vietti TJ, Gehan EA, et al. Ewing’s sarcoma metastatic at diagnosis. Results and comparisons of two intergroup Ewing’s sarcoma studies. Cancer. 1990;66(5):887–893. | |
Rodriguez-Galindo C, Liu T, Krasin MJ, et al. Analysis of prognostic factors in Ewing sarcoma family of tumors: review of St Jude Children’s Research Hospital studies. Cancer. 2007;110(2):375–384. | |
Miser JS, Krailo MD, Tarbell NJ, et al. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide – a Children’s Cancer Group and Pediatric Oncology Group study. J Clin Oncol. 2004;22(14):2873–2876. | |
Ahrens S, Hoffmann C, Jabar S, et al. Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol. 1999;32(3):186–195. | |
Oberlin O, Deley MC, Bui BN, et al. Prognostic factors in localized Ewing’s tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer. 2001;85(11):1646–1654. | |
Rodriguez-Galindo C, Billups CA, Kun LE, et al. Survival after recurrence of Ewing tumors: the St Jude Children’s Research Hospital experience, 1979–1999. Cancer. 2002;94(2):561–569. | |
Biermann JS. Updates in the treatment of bone cancer. J Natl Compr Canc Netw. 2013;11(5S):681–683. | |
Mankin HJ, Mankin CJ, Simon MA. The hazards of the biopsy, revisited. Members of the Musculoskeletal Tumor Society. J Bone Joint Surg Am. 1996;78(5):656–663. | |
Heudel PE, Cousin P, Lurkin A, et al. Territorial inequalities in management and conformity to clinical guidelines for sarcoma patients: an exhaustive population-based cohort analysis in the Rhône-Alpes region. Int J Clin Oncol. Epub August 10, 2013. | |
Judson I. Role of expert centres in the management of sarcomas. Eur J Cancer Suppl. 2013;11(2):310–311. | |
Gupta AA, Pappo A, Saunders N, et al. Clinical outcome of children and adults with localized Ewing sarcoma. Cancer. 2010;116(13):3189–3194. | |
Ahmed SK, Robinson SI, Okuno SH, Rose PS, Laack NN. Adult Ewing sarcoma: survival and local control outcomes in 102 patients with localized disease. Sarcoma. 2013;2013:681425. | |
Pieper S, Ranft A, Braun-Munzinger G, Jurgens H, Paulussen M, Dirksen U. Ewing’s tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-EWING 99. Onkologie. 2008;31(12):657–663. | |
Yock TI, Krailo M, Fryer CJ, et al. Local control in pelvic Ewing Sarcoma: analysis from INT-0091 – a report from the Children’s Oncology Group. J Clin Oncol. 2006;24(24):3838–3843. | |
Zogopoulos G, Teskey L, Sung L, et al. Ewing sarcoma: favourable results with combined modality therapy and conservative use of radiotherapy. Pediatr Blood Cancer. 2004;43(1):35–39. | |
Schuck A, Ahrens S, von Schorlemer I, et al. Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2005;63(5):1562–1567. | |
Raciborska A, Bilska K, Drabko K, et al. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer. 2013;60(10):1621–1625. | |
Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795–800. | |
Casey DA, Wexler LH, Merchant MS, et al. Irinotecan and temozolomide for Ewing sarcoma: The Memorial Sloan-Kettering experience. Pediatr Blood Cancer. 2009;53(6):1029–1034. | |
Wagner L, Turpin B, Nagarajan R, Weiss B, Cripe T, Geller J. Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr Blood Cancer. 2013;60(9):1447–1451. | |
Jones RL, Ferrari S, Blay JY, et al. A Phase II multicenter, open-label, clinical and pharmokinetic trial of PM00104 in patients with advanced Ewing family of tumors. Invest New Drugs. 2014;32(1):171–177. | |
Fox E, Aplenc R, Bagatell R, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. 2010;28(35):5174–5181. | |
Fox E, Patel S, Wathen JK, et al. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist. 2012;17(3):321. | |
Nesbit ME Jr, Gehan EA, Burgert EO Jr, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8(10):1664–1674. | |
Haeusler J, Ranft A, Boelling T, et al. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer. 2010;116(2):443–450. | |
Letourneau PA, Shackett B, Xiao L, et al. Resection of pulmonary metastases in pediatric patients with Ewing sarcoma improves survival. J Pediatr Surg. 2011;46(2):332–335. | |
Meyers PA, Krailo MD, Ladanyi M, et al. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing’s sarcoma does not improve prognosis. J Clin Oncol. 2001;19(11):2812–2820. | |
Ladenstein R, Pötschger U, Le Deley MC, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 Trial. J Clin Oncol. 2010;28(20):3284–3291. | |
Luksch R, Tienghi A, Hall KS, et al. Primary metastatic Ewing’s family tumors: results of the Italian Sarcoma Group and Scandinavian Sarcoma Group ISG/SSG IV study including myeloablative chemotherapy and total-lung irradiation. Ann Oncol. 2012;23(11):2970–2976. | |
Burdach S, van Kaick B, Laws HJ, et al. Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Dusseldorf University Medical Center, Germany and St Anna Kinderspital, Vienna, Austria. Ann Oncol. 2000;11(11):1451–1462. | |
Barrett D, Fish JD, Grupp SA. Autologous and allogeneic cellular therapies for high-risk pediatric solid tumors. Pediatr Clin North Am. 2010;57(1):47–66. | |
Lucas KG, Schwartz C, Kaplan J. Allogeneic stem cell transplantation in a patient with relapsed Ewing sarcoma. Pediatr Blood Cancer. 2008;51(1):142–144. | |
Meyer JS, Nadel HR, Marina N, et al. Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group Bone Tumor Committee. Pediatr Blood Cancer. 2008;51(2):163–170. | |
Kolb EA, Kushner BH, Gorlick R, et al. Long-term event-free survival after intensive chemotherapy for Ewing’s family of tumors in children and young adults. J Clin Oncol. 2003;21(18):3423–3430. | |
Strong LC, Herson J, Osborne BM, Sutow WW. Risk of radiation-related subsequent malignant tumors in survivors of Ewing’s sarcoma. J Natl Cancer Inst. 1979;62(6):1401–1406. | |
Ginsberg JP, Goodman P, Leisenring W, et al. Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst. 2010;102(16):1272–1283. | |
Saylors RL 3rd, Stine KC, Sullivan J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol. 2001;19(15):3463–3469. | |
Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol Cell Biol. 2004;24(16):7275–7283. | |
Scotlandi K, Benini S, Nanni P, et al. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res. 1998;58(18):4127–4131. | |
Martins AS, Mackintosh C, Martin DH, et al. Insulin-like growth factor I receptor pathway inhibition by ADW742, alone or in combination with imatinib, doxorubicin, or vincristine, is a novel therapeutic approach in Ewing tumor. Clin Cancer Res. 2006;12(11 Pt 1):3532–3540. | |
Manara MC, Landuzzi L, Nanni P, et al. Preclinical in vivo study of new insulin-like growth factor-I receptor-specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007;13(4):1322–1330. | |
Houghton PJ, Morton CL, Gorlick R, et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the pediatric preclinical testing program. Pediatr Blood Cancer. 2010;54(7):921–926. | |
Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29(34):4541–4547. | |
Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. | |
Juergens H, Daw NC, Geoerger B, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–4540. | |
Malempati S, Weigel B, Ingle AM, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(3):256–262. | |
Kolb EA, Gorlick R, Maris JM, et al. Combination testing (stage 2) of the Anti-IGF-1 receptor antibody IMC-A12 with rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58(5):729–735. | |
O’Neill A, Shah N, Zitomersky N, et al. Insulin-like growth factor 1 receptor as a therapeutic target in Ewing sarcoma: lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature. Sarcoma. 2013;2013:450478. | |
Gorlick R, Janeway K, Lessnick S, Randall RL, Marina N. Children’s Oncology Group’s 2013 blueprint for research: bone tumors. Pediatr Blood Cancer. 2013;60(6):1009–1015. | |
Mateo-Lozano S, Tirado OM, Notario V. Rapamycin induces the fusion-type independent downregulation of the EWS/FLI-1 proteins and inhibits Ewing’s sarcoma cell proliferation. Oncogene. 2003;22(58):9282–9287. | |
Houghton PJ, Morton CL, Kolb EA, et al. Initial testing (stage 1) of the mTOR inhibitor rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50(4):799–805. | |
Houghton PJ, Morton CL, Gorlick R, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the pediatric preclinical testing program. Mol Cancer Ther. 2010;9(1):101–112. | |
Bagatell R, Norris R, Ingle AM, et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer. 2014;61(5):833–839. | |
Brenner JC, Ateeq B, Li Y, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19(5):664–678. | |
Brenner JC, Feng FY, Han S, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s Sarcoma. Cancer Res. 2012;72(7):1608–1613. | |
Norris RE, Adamson PC, Nguyen VT, Fox E. Preclinical evaluation of the PARP inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatr Blood Cancer. 2014;61(1):145–150. | |
Baker VV, Shingleton HM, Hatch KD, Miller DM. Selective inhibition of c-myc expression by the ribonucleic acid synthesis inhibitor mithramycin. Am J Obstet Gynecol. 1988;158(4):762–767. | |
Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest. 1991;88(5):1613–1621. | |
Koutsodontis G, Kardassis D. Inhibition of p53-mediated transcriptional responses by mithramycin A. Oncogene. 2004;23(57):9190–9200. | |
Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42(27):8313–8324. | |
Snyder RC, Ray R, Blume S, Miller DM. Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry. 1991;30(17):4290–4297. | |
Kofman S, Medrek TJ, Alexander RW. Mithramycin in the treatment of embryonal cancer. Cancer. 1964;17:938–948. | |
Kofman S, Perlia CP, Economou SG. Mithramycin in the treatment of metastatic Ewing’s sarcoma. Cancer. 1973;31(4):889–893. | |
Grohar PJ, Woldemichael GM, Griffin LB, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J Natl Cancer Inst. 2011;103(12):962–978. | |
Erkizan HV, Kong Y, Merchant M, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15(7):750–756. | |
Barber-Rotenberg JS, Selvanathan SP, Kong Y, et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget. 2012;3(2):172–182. | |
Erkizan HV, Scher LJ, Gamble SE, et al. Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell Cycle. 2011;10(19):3397–3408. | |
Howlader N, Noone A, Krapcho M, et al. SEER cancer statistics review, 1975–2008. Available from: http://seer.cancer.gov/csr/1975_2008. Accessed November 23, 2013. | |
Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir GM. Chromosome study of Ewing’s sarcoma (ES) cell lines. Consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet Cytogenet. 1984;12(1):1–19. | |
Kaneko Y, Yoshida K, Handa M, et al. Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosomes Cancer. 1996;15(2):115–121. | |
Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 2003;63(15):4568–4576. | |
Ng TL, O’Sullivan MJ, Pallen CJ, et al. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn. 2007;9(4):459–463. | |
Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PC. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15(7):2259–2268. | |
Yamaguchi S, Yamazaki Y, Ishikawa Y, Kawaguchi N, Mukai H, Nakamura T. EWSR1 is fused to POU5F1 in a bone tumor with translocation t(6;22)(p21;q12). Genes Chromosomes Cancer. 2005;43(2):217–222. | |
Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol. 2011;24(3):333–342. | |
Mastrangelo T, Modena P, Tornielli S, et al. A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene. 2000;19(33):3799–3804. | |
Wang L, Bhargava R, Zheng T, et al. Undifferentiated small round cell sarcomas with rare EWS gene fusions: identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn. 2007;9(4):498–509. | |
Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15(13):2125–2137. | |
Rosito P, Mancini AF, Rondelli R, et al. Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone. Cancer. 1999;86(3):421–428. | |
Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin Oncol. 2001;19(6):1818–1829. | |
Obata H, Ueda T, Kawai A, et al. Clinical outcome of patients with Ewing sarcoma family of tumors of bone in Japan. Cancer. 2007;109(4):767–775. | |
Granowetter L, Womer R, Devidas M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group study. J Clin Oncol. 2009;27(15):2536–2541. | |
Ferrari S, Sundby Hall K, Luksch R, et al. Nonmetastatic Ewing family tumors: high-dose chemotherapy with stem cell rescue in poor responder patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III protocol. Ann Oncol. 2011;22(5):1221–1227. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.