")
Back to Journals » OncoTargets and Therapy » Volume 8
Apoptotic effect of cordycepin combined with cisplatin and/or paclitaxel on MA-10 mouse Leydig tumor cells
Authors Kang F , Chen P, Pan B, Lai M, Chen Y , Huang B , Huang M
Received 20 April 2015
Accepted for publication 24 July 2015
Published 1 September 2015 Volume 2015:8 Pages 2345—2360
DOI https://doi.org/10.2147/OTT.S87010
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Faris Farassati
Fu-Chi Kang,1 Pei-Jung Chen,2 Bo-Syong Pan,2,3 Meng-Shao Lai,2,3 Yung-Chia Chen,4 Bu-Miin Huang2,3
1Department of Anesthesia, Chi Mei Medical Center, Chiali, 2Department of Cell Biology and Anatomy, 3Institute of Basic Medical Sciences, College of Medicine, National Cheng Kung University, Tainan, 4Department of Anatomy, School of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan, Republic of China
Background: Chemotherapy is not limited to a single treatment, and the evidence demonstrates that different drug combinations can have positive results in patients. In this study, we sought to determine whether cordycepin combined with cisplatin and/or paclitaxel would have an additive effective on inducing apoptosis in mouse Leydig tumor cells, and the mechanisms were also briefly examined.
Methods: The additive effects of cordycepin combined with cisplatin and/or paclitaxel on apoptosis in MA-10 cells were investigated by monitoring changes in morphological characteristics and examining cell viability, flow cytometry assays, and Western blot analyses.
Results: Combination of cordycepin plus cisplatin and/or paclitaxel for 12 and 24 hours induced apoptotic features in MA-10 cells. The MTT assay showed that the combination treatment reduced the viability of MA-10 cells in a dose-dependent manner, with additive effects. Cell cycle analysis showed that combination treatment significantly increased subG1 phase cell numbers in MA-10 cells, indicating apoptosis. Moreover, cordycepin plus cisplatin and/or paclitaxel significantly induced cleavage of caspase-8, caspase-9, caspase-3, and poly ADP-ribose polymerase, and phosphorylation of c-Jun NH2-terminal kinase, extracellular signal-regulated kinase, p38, and p53 proteins in MA-10 cells.
Conclusion: Cordycepin plus cisplatin and/or paclitaxel can have an additive effect on apoptosis in MA-10 cells, with activation of caspase, mitogen-activated protein kinase, and p53 signal pathways.
Keywords: cordycepin, cisplatin, paclitaxel, apoptosis, drug combination, additive effect, MA-10 cells, Leydig tumor cells
Introduction
Leydig cells produce testosterone, which is the major androgenic steroid circulating in blood.1 Testosterone is essential for correct development of the male reproductive system during puberty. Any disorder of the hypothalamic-pituitary-testis axis might cause abnormal steroid secretion, which could result in oncogenesis.2 Testicular cancer, which includes germ cell, Sertoli cell, and Leydig cell tumors, is one of the cancers most commonly diagnosed in men aged 15–35 years, with approximately 8,000 cases detected in the USA annually.3 Surgery, radiation, and chemotherapy have been used to treat testicular cancer, but may cause organ failure and skin irritation. Although chemotherapy might be beneficial for patients, it has side effects and resistance.4 Due to the disadvantages of treatment with a single chemotherapeutic agent, drug combinations at lower doses may increase efficacy and decrease side effects and resistance in patients.
Studies have demonstrated that combination therapy of paclitaxel and/or cisplatin with medicinal herbs, such as beta-elemene (a novel plant-derived antineoplastic agent with low toxicity), could have better efficacy, significantly increasing the cytotoxicity of cisplatin in androgen-independent DU145 and PC-3 prostate carcinoma cell lines.5 Also, the use of plant compounds, such as perillyl alcohol or methyl jasmonate, in combination with anticancer drugs did improve their efficacy as inhibitors of cancer cell growth and induce cell apoptosis.6 Further, paclitaxel has a wide range of synergistic antitumor effects when used in combination with other chemotherapeutic agents, such as 5-fluorouracil or cisplatin.7
Cordycepin, a compound isolated from Cordyceps sinensis, has been shown to have antitumor effects.8–11 Cordycepin has been reported to inhibit formation of polyadenylate polymerase and to inactivate mRNA polyadenylation and induce apoptosis of tumor cells.12,13 Paclitaxel, an extract from the bark of the Pacific yew tree (Taxus brevifolia), was first isolated in 1963, and can induce cell death by disrupting the microtubular dynamics involved in cell mitosis and proliferation.14,15 Paclitaxel has been used to treat breast, ovarian, lung, and head and neck cancers. Cisplatin, also known as cis-diamminedichloroplatinum(II), is widely used for the treatment of malignancies, including testicular, ovarian, bladder, and head and neck cancers.16,17 Cisplatin acts by binding to nuclear DNA and subsequently interfering with normal transcription and/or DNA replication, which induces death of tumor cells by apoptosis.18
In apoptosis, there are two major signaling pathways, ie, the death receptor pathway (extrinsic caspase) and the mitochondrial pathway (intrinsic caspase).19,20 In terms of their function, caspases can be divided into two groups, ie, initiator caspases, including caspase-8, caspase-9, and caspase-10, and effector caspases, including caspase-3, caspase-6, and caspase-7. Initiator caspases are responsible for cleaving and activating effector caspases.21 The cleavage of caspases will further cleave poly ADP-ribose polymerase (PARP), resulting in cell death.22 It has been shown that apoptosis is also regulated by mitogen-activated protein kinase (MAPK), which consists of three family membranes, extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 proteins.23 Moreover, a study has demonstrated that the p53 pathway plays an essential role in regulating cell cycle arrest related to apoptosis.24
We have demonstrated that cordycepin significantly activates adenosine subtype receptors, the caspase pathway, and cell cycle arrest to induce apoptotic death in MA-10 mouse Leydig cells.9,10 Studies have shown that cordycepin, paclitaxel, and cisplatin all have antitumor effects,9,10,14,18 and cordycepin in combination with cisplatin had a synergistic antitumor effect in human oral cancer cells.25,26 In the present study, we attempted to clarify the effects of combination treatment with cordycepin, paclitaxel, and/or cisplatin on MA-10 cell apoptosis and to determine the mechanism of action involved. We found that cordycepin plus paclitaxel and/or cisplatin had additive effects on apoptosis in MA-10 cells, with an increase in subG1 cells and activation of the caspase, MAPK, and p53 pathways. These findings could encourage the development of more effective regimens using concomitant chemotherapeutic agents in lower doses, thereby reducing side effects and resistance in testicular cancers.
Materials and methods
Chemicals
Cordycepin, paclitaxel, cisplatin, Waymouth MB 752/1 medium, propidium iodide, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), RNase A, bovine serum albumin, and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemicals (St Louis, MO, USA). Fetal bovine serum, lyophilized trypsin-ethylenediaminetetraacetic acid (EDTA), Dulbecco’s phosphate-buffered saline (PBS), and gentamicin sulfate were purchased from Gibco (Grand Island, NY, USA). Sodium hydroxide, hydrochloric acid, sodium dodecyl sulfate, EDTA, isopropyl alcohol, chloroform, and Tween-20 were purchased from Merck (Darmstadt, Germany). Acrylamide was purchased from JT Baker (Phillipsburg, NJ, USA). HEPES was purchased from Mallinckrodt Baker Inc (Paris, KT, USA). Sodium bicarbonate, sodium carbonate, and sodium chloride were purchased from Riedel-de Haën (Seelze, Germany). Antibodies against cleaved caspase-3, caspase-8, and caspase-9, phospho-ERK1/2, phosphor-SAPK/JNK, phosphor-p38, ERK1/2, SAPK/JNK, and p38 were purchased from Cell Signaling (Beverly, MA, USA). Tris base, Z-LEHD-FMK (caspase-9 inhibitor), Z-IETD-FMK (caspase-8 inhibitor), Z-DEVD-FMK (caspase-3 inhibitor), and β-actin were purchased from Calbiochem (San Diego, CA, USA). Anti-PARP antibody was purchased from Oncogene (San Diego, CA, USA).
Morphological study
The MA-10 cell line was a gift from Dr Mario Ascoli (University of Iowa, Iowa City, IA, USA) and was maintained at 37°C in a humidified environment containing 95% air and 5% CO2 for all experiments. MA-10 cells were seeded in 96-well plates that contained 1.5×104 cells and 100 μL of serum-free medium. After reaching 70%–80% confluence, the cells were treated with serum-free medium containing cordycepin (100 μM), paclitaxel (50 nM), cisplatin (100 μM), or cordycepin (100 μM) plus cisplatin (100 μM) and/or paclitaxel (50 nM) followed by incubation for 24 hours. The morphology of the cells was then observed under a CK40 light microscope (Olympus, Tokyo, Japan). Apoptosis is characterized by loss of contact between cells and the matrix, along with blebbing of the plasma membrane.27
MTT cytotoxicity assay
MA-10 cells were seeded in 96-well plates in quadruplicate, which contained 2.5×104 cells and 100 μL of serum-free medium. After reaching 70%–80% confluence, the cells were treated with serum-free medium containing cordycepin (100 μM), paclitaxel (50 nM), cisplatin (100 μM), or cordycepin (100 μM) plus cisplatin (100 μM) and/or paclitaxel (50 nM) and incubated for 24 hours. MTT was then added to the wells (to a final concentration of 0.5 mg/mL) for conversion by mitochondrial dehydrogenases into color-dense formazan crystals in viable cells. Four hours later, DMSO was added to the wells to dissolve the formazan. Quadruplicate DMSO solutions were mixed and absorbance was measured at 590 nm using an enzyme-linked immunosorbent assay reader (Opsys MR™, Dynex Technologies Inc, Chantilly, VA, USA).
Apoptosis analysis by flow cytometry
DNA fragmentation was assessed by flow cytometry using propidium iodide. MA-10 cells (6.5×105) were seeded in a 6 cm dish with 2 mL of serum medium. After reaching 70%–80% confluence, the cells were treated with serum-free medium containing cordycepin (100 μM), paclitaxel (50 nM), cisplatin (100 μM), or cordycepin (100 μM) plus cisplatin (100 μM) and/or paclitaxel (50 nM) and incubated for 24 hours. Cells floating in the medium were then collected, and the attached cells were trypsinized (trypsin-EDTA 0.5%–5.35% mM in PBS) and washed with PBS. After centrifugation, the cell pellets were washed with FACS buffer and fixed in 70% ethanol for 2 hours at −20°C. The cell pellets were next washed in cold FACS buffer to remove the ethanol, and then centrifuged and resuspended in staining solution containing RNase A (100 μg/mL in PBS) and propidium iodide (40 μg/mL in PBS). After staining, the cells were investigated by flow cytometry, with excitation set at 469 nm on FACScan using CellQuest software (Becton Dickinson, Mountain View, CA, USA).
Immunoblot analysis
After the various treatments, the cells were lysed and protein extraction was performed. The protein concentration in the cell lysates were determined by the Lowry method.28 Cell proteins (30 μg) were separated in 12% sodium dodecyl sulfate-polyacrylamide gel, which was performed at 90 V for 3 hours using standard running buffer (24 mM Tris-HCl, 0.19 M glycine, 0.5% sodium dodecyl sulfate, pH 8.3), and electrophoretically transferred to a polyvinylidene difluoride membrane at 400 mA for 90 minutes in transfer buffer (20 mM Tris-HCl, 150 mM glycine, 10% methanol, 0.01% sodium dodecyl sulfate). The membranes were blocked with 5% non-fat milk, washed, and subsequently incubated with primary antibodies at a 1:2,000 dilution for 16–18 hours at 4°C. After washing, the membrane was incubated with horseradish peroxidase-conjugated goat antimouse antibody or goat antirabbit antibody, and then visualized using an enhanced chemiluminescence detection kit (Amersham-Pharmacia International PLC, Little Chalfont, UK). The optical density of each protein band was quantified using a Quantity One (PDI, Huntington Station, NY, USA) computer-assisted image analysis system.29 The amount of β-actin and total MAPK protein in each lane was also detected as a control.
Statistical analysis
Each datum point in the figures represents the mean ± standard error of the mean of three or four separate experiments. Statistically significant differences between treatments and control were determined by one-way analysis of variance and then Tukey’s test. Statistical significance was set at P<0.05.
Results
Effects of cordycepin + paclitaxel and/or cisplatin on morphology and viability in MA-10 cells
MA-10 cells were treated with 100 μM cordycepin, 50 nM paclitaxel, or 100 μM cisplatin alone or in combination with cordycepin + paclitaxel and/or cisplatin without serum for 24 hours. The changes in morphology were then observed under a light microscope. Cells in the control and in DMSO demonstrated a normal polygonal shape, with firm attachment to the culture dishes, which is a normal cell growth phenomenon (Figure 1A and B). After 24 hours, cells treated with 100 μM cordycepin, 50 nM paclitaxel, or 100 μM cisplatin had a rounded-up appearance but still adhered to the matrix (Figure 1C–E). Treatment for 24 hours with combinations of cisplatin + cordycepin, cisplatin + paclitaxel, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel resulted in more loss of cell attachment to the matrix, more membrane blebbing, and many more floating cells (Figure 1F–I). The morphological study suggested that the combination treatments induced more cell death in MA-10 cells than treatment with cordycepin, paclitaxel, or cisplatin alone.
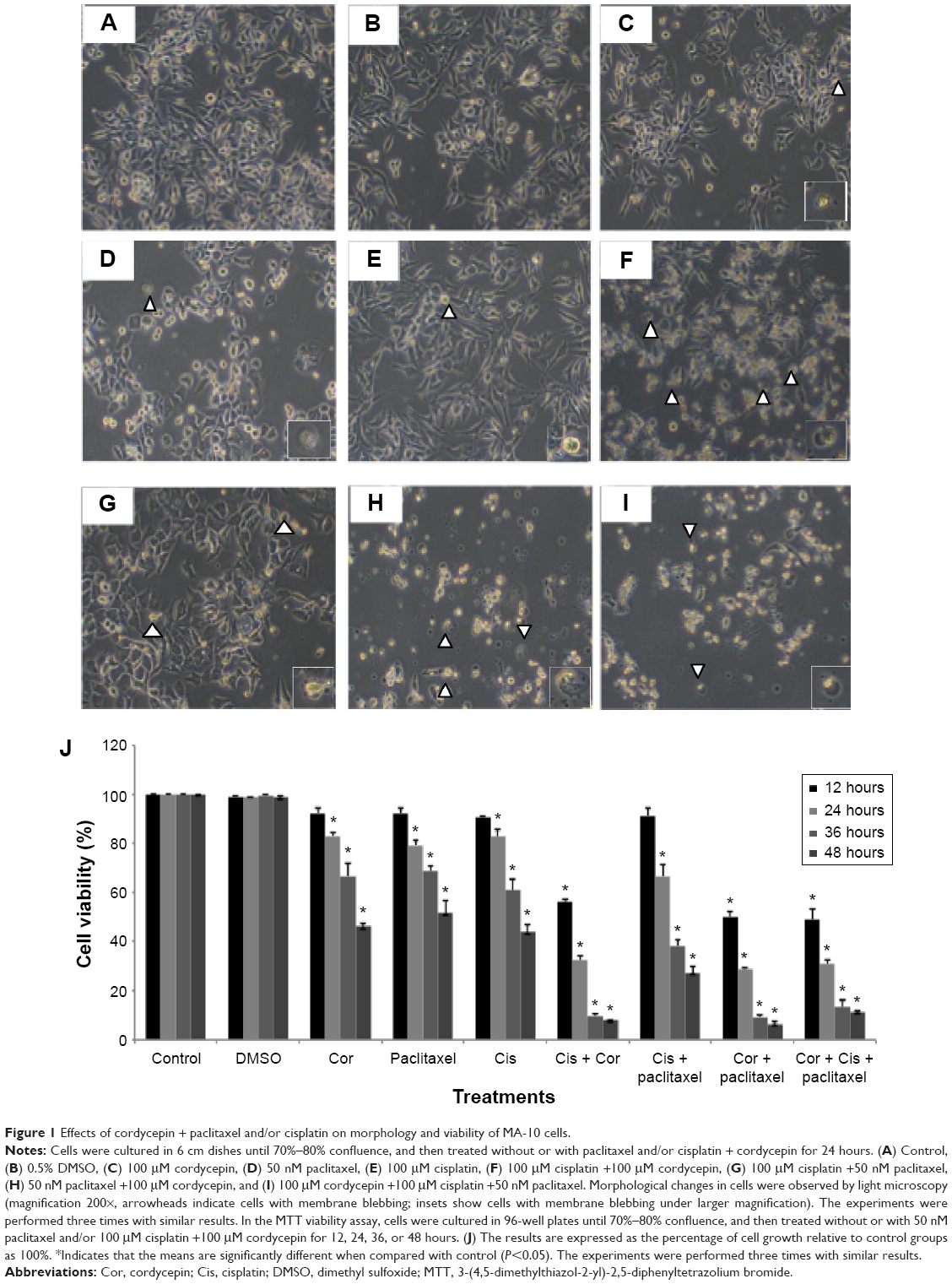 | Figure 1 Effects of cordycepin + paclitaxel and/or cisplatin on morphology and viability of MA-10 cells. |
An MTT assay was used to further examine the effect of cordycepin + paclitaxel and/or cisplatin on the viability of MA-10 cells. Cell viability was not significantly decreased by treatment with cordycepin, paclitaxel, cisplatin, or cisplatin + paclitaxel, but was reduced to 56.02% by cisplatin + cordycepin, 50.28% by cordycepin + paclitaxel, and 49.06% by cordycepin + cisplatin + paclitaxel after 12 hours of treatment (Figure 1J). After 24 hours of treatment, cell viability was reduced to 83.00% by cordycepin, 79.29% by paclitaxel, 82.66% by cisplatin, 32.41% by cordycepin + cisplatin, 66.61% by cisplatin + paclitaxel, 28.61% by cordycepin + paclitaxel, and 30.91% by cordycepin + cisplatin + paclitaxel (Figure 1J). After 36 hours of treatment, cell viability was reduced to 66.63% by cordycepin, 68.52% by paclitaxel, 60.81% by cisplatin, 9.76% by cordycepin + cisplatin, 38.20% by cisplatin + paclitaxel, 9.34% by cordycepin + paclitaxel, and 13.20% by cordycepin + cisplatin + paclitaxel (Figure 1J). After 48 hours of treatment, cell viability was reduced to 46.12% by cordycepin; 51.75% at paclitaxel, 44.01% by cisplatin, 8.16% by cordycepin + cisplatin, 27.36% by cisplatin + paclitaxel, 6.59% by cordycepin + paclitaxel, and 11.50% by cordycepin + cisplatin + paclitaxel (Figure 1J). These results demonstrate that cordycepin combined with paclitaxel and/or cisplatin significantly reduced cell viability more than treatment with each drug alone and with an additive apoptotic effect.
Effects of cordycepin + paclitaxel and/or cisplatin on the cell cycle in MA-10 cells
To investigate whether cordycepin combined with paclitaxel and/or cisplatin could induce apoptosis, MA-10 cells were examined for DNA content by propidium iodide staining and flow cytometry analysis. A feature of cell apoptosis is DNA fragmentation of genomic DNA into multiples of 180–200 bp units. In the absence of drugs, cells can be divided into two phases, ie, the G1 (diploid) phase and G2/M phase, with greater numbers of cell accumulation in G1 phase than G2/M phase. In the presence of 100 μM cordycepin, 50 nM paclitaxel, but not cisplatin, and all the combination treatments for 24 hours, the percentage of abnormal DNA content in subG1 phase (haploid), increased (Figure 2A). Figure 2B demonstrates that the percentage of control cells in subG1 phase was approximately 5% and was increased significantly to 53.76% by cordycepin, 19.65% by paclitaxel, 53.53% by cisplatin + cordycepin, 18.34% by cisplatin + paclitaxel, 53.43% by cordycepin + paclitaxel, and 57.79% by cordycepin + cisplatin + paclitaxel after treatment for 24 hours (P<0.05). There was no difference between the control and cisplatin groups (Figure 2B). Figure 2C demonstrated that the percentage of control cells in G1 phase was approximately 70%, and was decreased significantly to 32.86% by cordycepin, 60.15% by paclitaxel, 58.76% by cisplatin, 30.2% by cisplatin + cordycepin, 37.12% by cisplatin + paclitaxel, 33.17% by cordycepin + paclitaxel, and 31.83% by cordycepin + cisplatin + paclitaxel after treatment for 24 hours (P<0.05). Figure 2D demonstrates that the percentage of control cells in G2/M phase was approximately 20%, which was increased significantly to 34.73% by cisplatin and 44.57% by cisplatin + paclitaxel after treatment for 24 hours (P<0.05). These results demonstrate that cordycepin combined with paclitaxel and/or cisplatin could induce apoptosis in MA-10 cells.
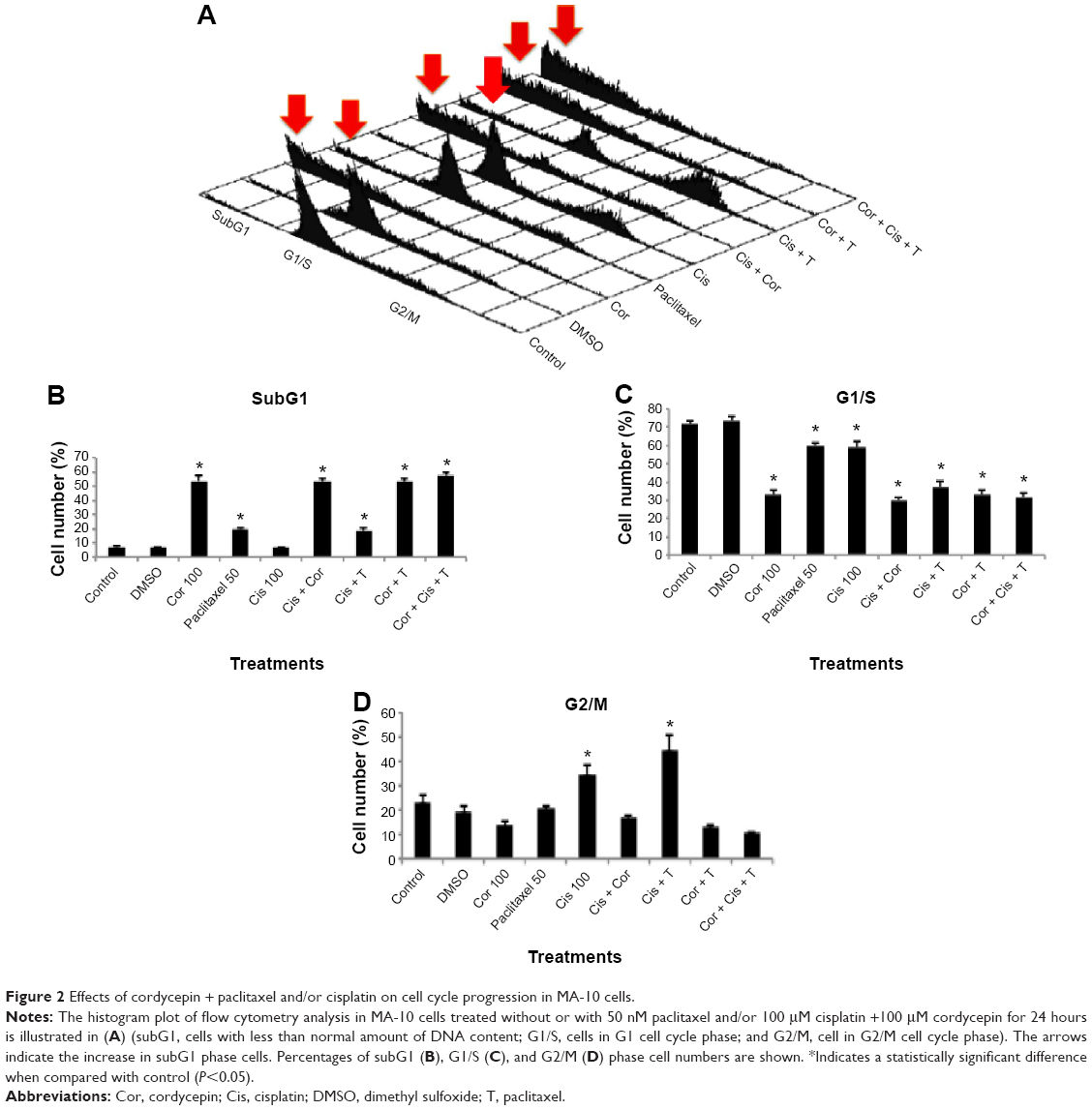 | Figure 2 Effects of cordycepin + paclitaxel and/or cisplatin on cell cycle progression in MA-10 cells. |
Effect of cordycepin + paclitaxel and/or cisplatin on cleavage of caspases and PARP proteins in MA-10 cells
We then investigated whether cordycepin + paclitaxel and or cisplatin could activate the caspase cascade to induce apoptosis in MA-10 cells. Treatment with cordycepin alone for 12 or 24 hours significantly increased the expression of cleaved caspase-8 (P<0.05, Figure 3A). The combinations of cisplatin + cordycepin, cisplatin + paclitaxel, cordycepin + paclitaxel, and cisplatin + cordycepin + paclitaxel for 12 and 24 hours also induced significant caspase-8 cleavage (P<0.05, Figure 3A). Treatment with cordycepin (100 μM), paclitaxel (50 nM), cisplatin (100 μM), cisplatin + paclitaxel, cordycepin + paclitaxel, cisplatin + cordycepin, and cordycepin + cisplatin + paclitaxel for 12 and 24 hours all induced significant caspase-9 cleavage compared to the control (P<0.05). Indeed, treatment with cisplatin + cordycepin for 12 hours had a synergistic effect on cleavage of caspase-9. Cordycepin (100 μM), paclitaxel (50 nM), cisplatin (100 μM), cisplatin + paclitaxel, cordycepin + paclitaxel, cisplatin + cordycepin, and cordycepin + cisplatin + paclitaxel, but not cisplatin alone, induced significant caspase-3 cleavage compared with control after 24 hours of treatment (P<0.05, Figure 3C). Significant cleavage of PARP was observed after treatment with cordycepin for 12 and 24 hours, paclitaxel for 12 and 24 hours, cisplatin for 24 hours, cisplatin + cordycepin for 12 hours, cordycepin + paclitaxel for 12 and 24 hours, and cordycepin + cisplatin + paclitaxel for 12 and 24 hours (P<0.05, Figure 3D). These results indicate that cordycepin combined with paclitaxel and/or cisplatin can induce the intrinsic and extrinsic apoptotic pathways by activating cleavage of caspase-8, caspase-9, caspase-3, and PARP proteins.
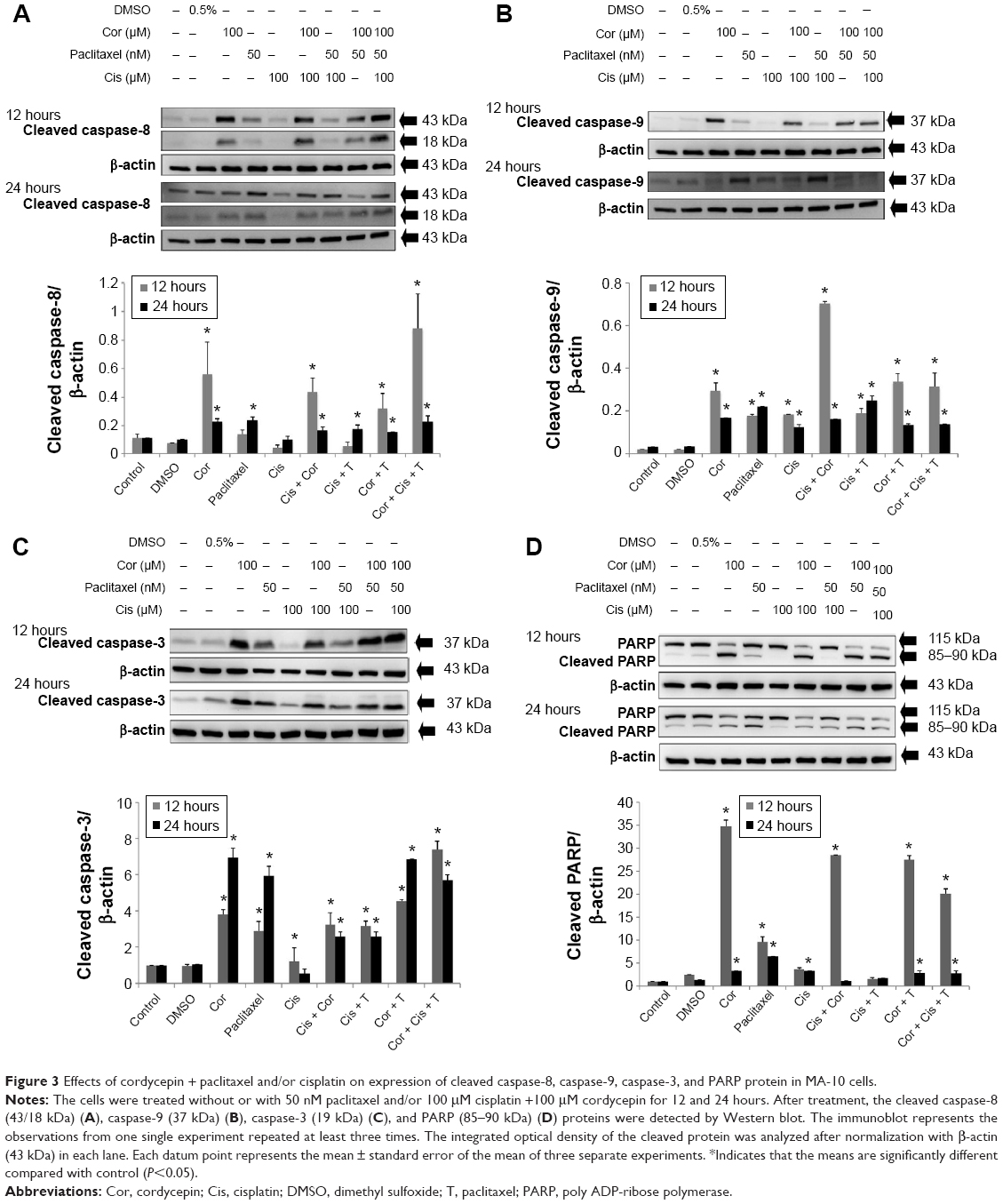 | Figure 3 Effects of cordycepin + paclitaxel and/or cisplatin on expression of cleaved caspase-8, caspase-9, caspase-3, and PARP protein in MA-10 cells. |
Effects of cordycepin + paclitaxel and/or cisplatin on MAPK proteins in MA-10 cells
Previous studies have shown that activation of MAPK is involved in apoptosis induced by cordycepin, cisplatin, and paclitaxel in various tumor cells.30–32 Therefore, we investigated whether the MAPK pathway was involved in apoptosis stimulated by cordycepin + paclitaxel and/or cisplatin in MA-10 cells. Treatment with cordycepin for 12 and 24 hours, paclitaxel for 24 hours, cisplatin for 24 hours, cisplatin + cordycepin for 12 and 24 hours, cisplatin + paclitaxel for 24 hours, cordycepin + paclitaxel for 12 and 24 hours, and cordycepin + cisplatin + paclitaxel for 12 and 24 hours all significantly induce expression of phosphorylated JNK in MA-10 cells (P<0.05, Figure 4A). Only treatment with cordycepin, cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel for 12 hours could induce significant expression of phosphorylated ERK in MA-10 cells (P<0.05, Figure 4B). Only treatment with cordycepin, cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel for 24 hours could induce significant expression of phosphorylated p38 in MA-10 cells (P<0.05, Figure 4C). These results indicate that cordycepin + paclitaxel and/or cisplatin could stimulate phosphorylation of the JNK, ERK1/2, and p38 MAPK pathways to induce apoptosis in MA-10 cells.
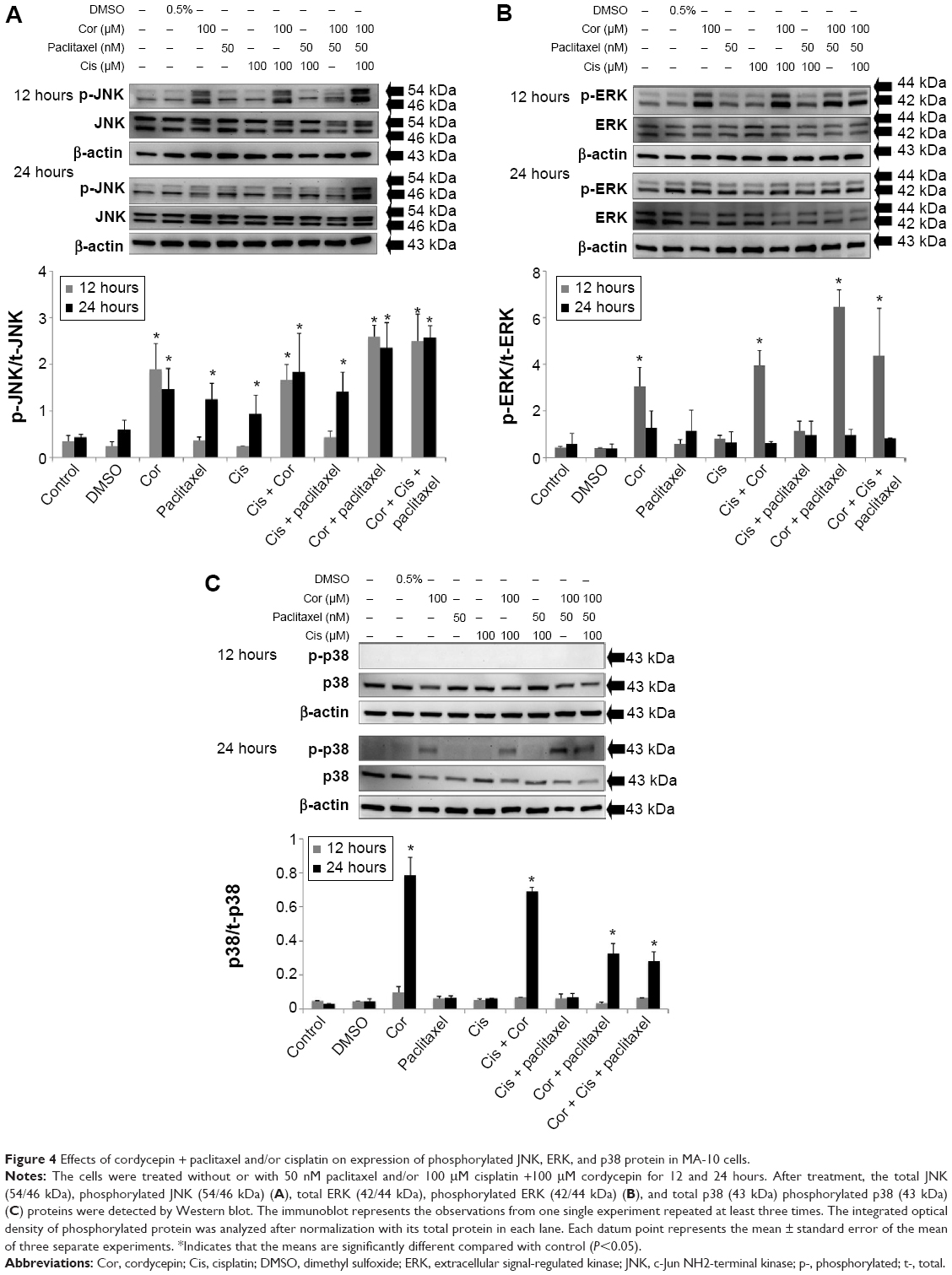 | Figure 4 Effects of cordycepin + paclitaxel and/or cisplatin on expression of phosphorylated JNK, ERK, and p38 protein in MA-10 cells. |
Inhibitors reversed phosphorylation of MAPK proteins induced by cordycepin + paclitaxel and/or cisplatin in MA-10 cells
To reconfirm whether phosphorylation of JNK, ERK, and p38 proteins could be induced by cordycepin + paclitaxel and/or cisplatin in MA-10 cells, inhibitors of JNK, ERK, and p38 (SP600125, PD98059, and SB203580, respectively) were used to abolish expression of phosphorylated JNK, ERK, and p38. SP600125 at 10, 50, and 100 μM significantly inhibited phosphorylation of JNK induced by cordycepin 100 μM (P<0.05, Figure 5A). In the combination experiments, SP600125 10 μM significantly reduced the expression of phosphorylated JNK after treatment with cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel for 12 hours (P<0.05, Figure 5B). PD98059 at 5, 10, 25, and 50 μM significantly inhibited phosphorylation of ERK induced by cordycepin 100 μM (P<0.05, Figure 6A). In the combination experiments, PD98059 5 μM significantly reduced the expression of phosphorylated ERK in the cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel groups treated for 12 hours (P<0.05, Figure 6B). SB203580 at 1, 5, and 10 μM significantly inhibited the phosphorylation of p38 induced by 100 μM cordycepin (P<0.05, Figure 7A). In the combination experiments, SB203580 10 μM significantly reduced the expression of phosphorylated p38 in the cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel groups (P<0.05, Figure 7A and B). These results again showed that cordycepin + paclitaxel and/or cisplatin could stimulate activation of the JNK, ERK1/2, and/or p38 MAPK pathways to induce apoptosis of MA-10 cells.
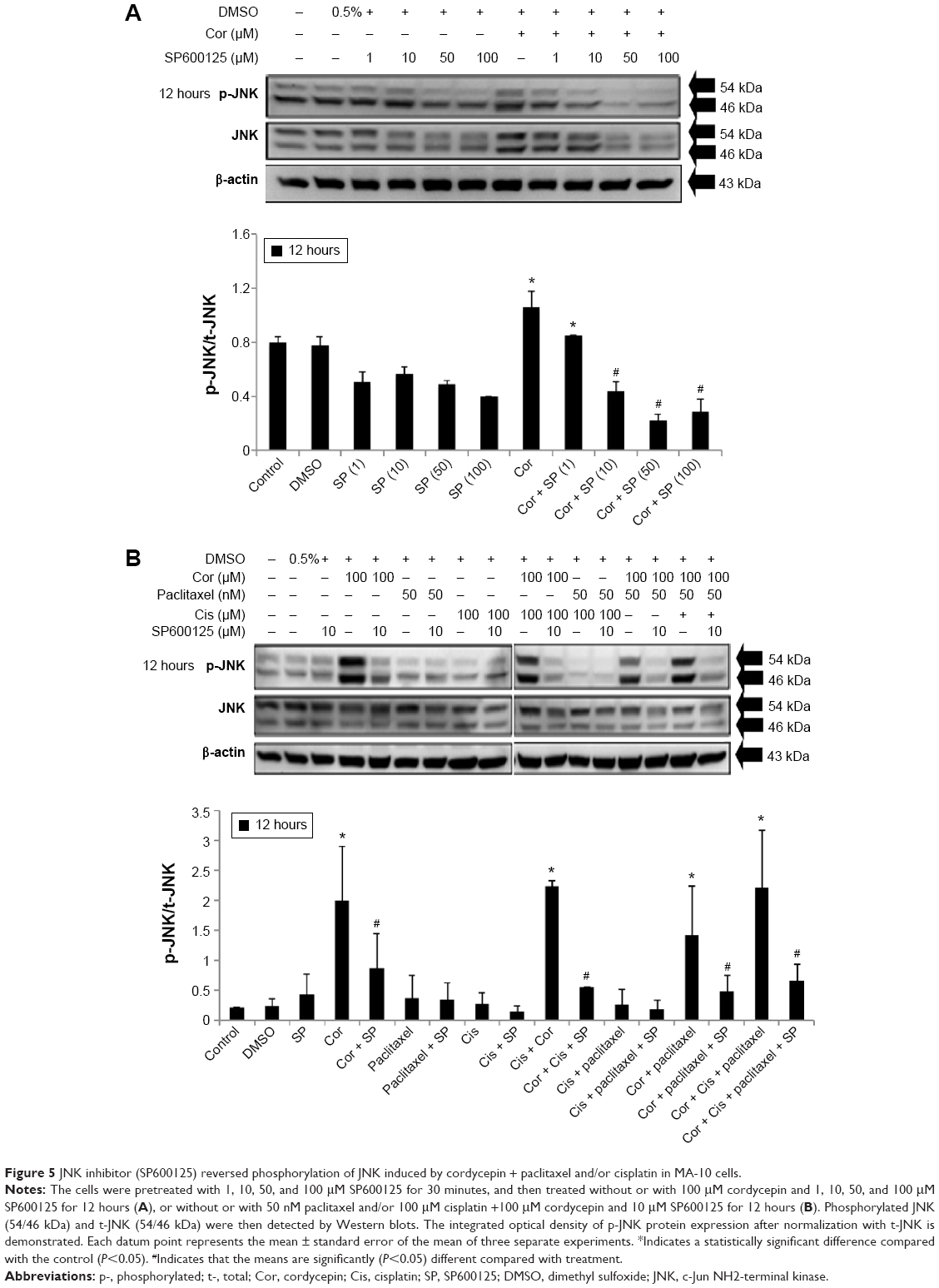 | Figure 5 JNK inhibitor (SP600125) reversed phosphorylation of JNK induced by cordycepin + paclitaxel and/or cisplatin in MA-10 cells. |
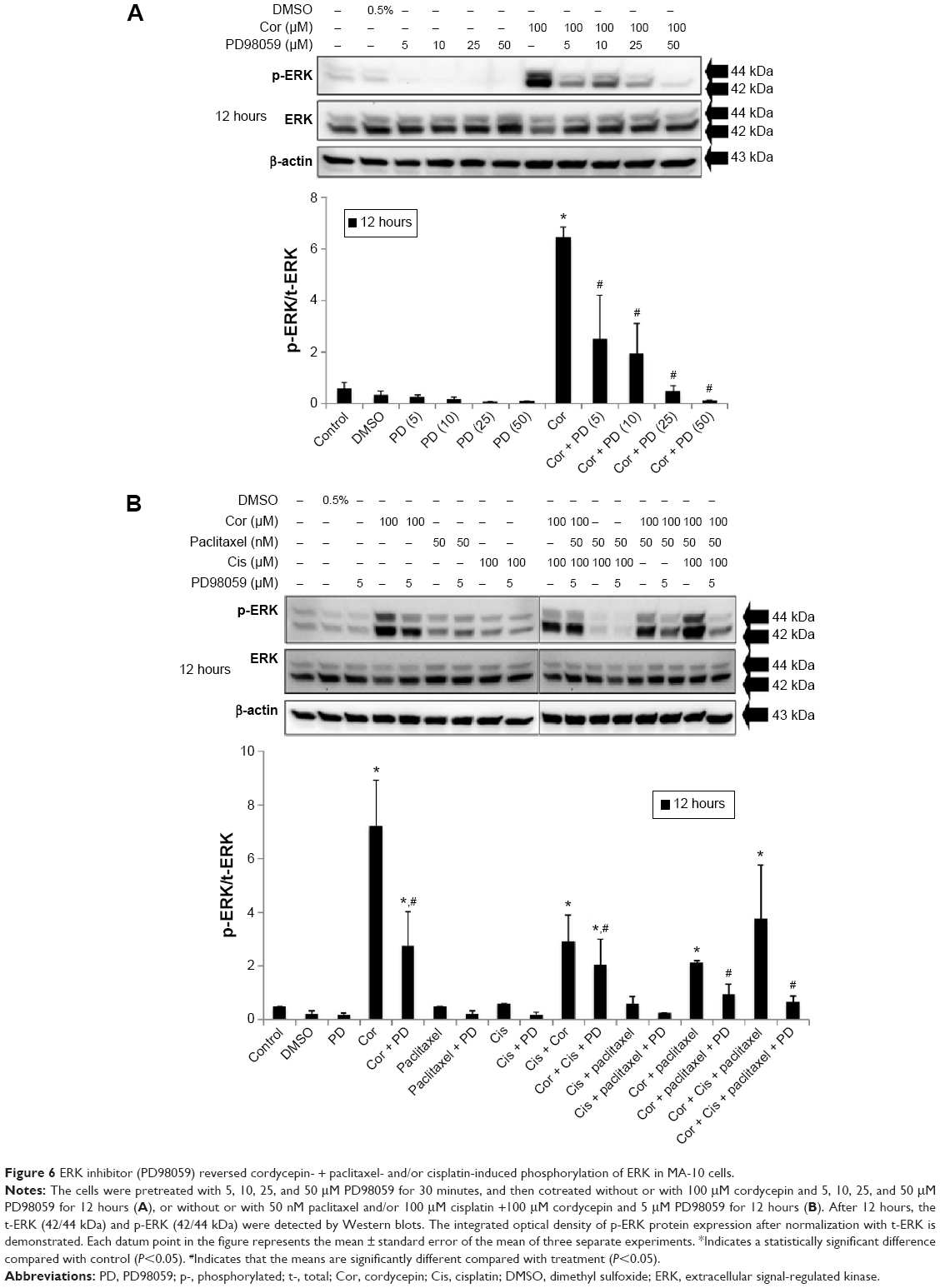 | Figure 6 ERK inhibitor (PD98059) reversed cordycepin- + paclitaxel- and/or cisplatin-induced phosphorylation of ERK in MA-10 cells. |
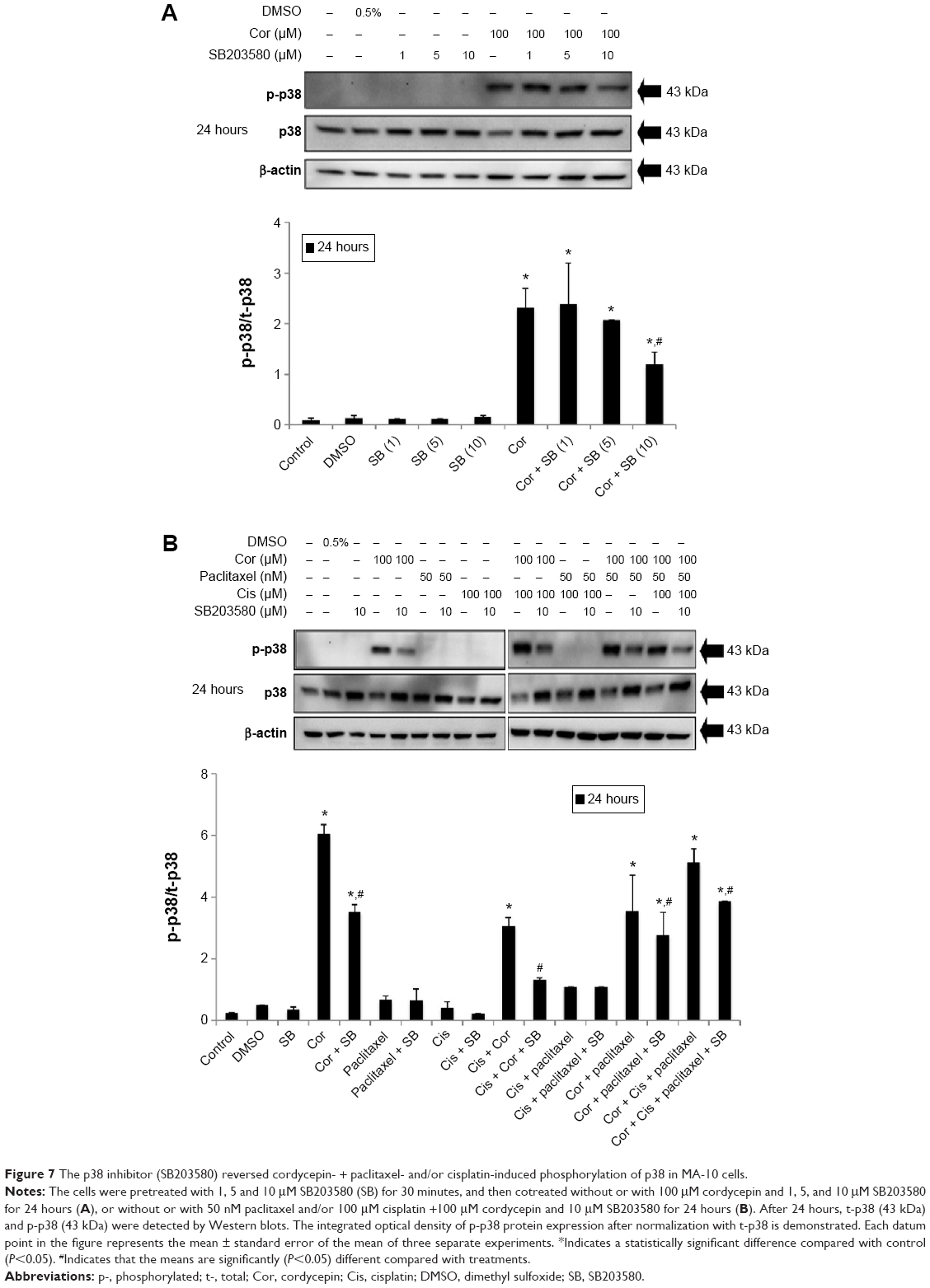 | Figure 7 The p38 inhibitor (SB203580) reversed cordycepin- + paclitaxel- and/or cisplatin-induced phosphorylation of p38 in MA-10 cells. |
Effects of cordycepin + paclitaxel and/or cisplatin on phosphorylation of p53 protein in MA-10 cells
The tumor suppressor p53 is the central player in a network protecting higher eukaryotic cells against various types of damage that might lead to genetic alterations.33 In addition, p53 protein can functionally interact with MAPK pathways.34 Moreover, studies have demonstrated that p53 can be induced by two widely used anticancer agents, ie, cisplatin and paclitaxel.35 Hence, we investigated whether expression of p53 protein was involved in apoptosis of MA-10 cells activated by cordycepin + paclitaxel and/or cisplatin. Expression of phosphorylated p53 protein in MA-10 cells treated with control and DMSO was low (Figure 8A and B). After 12 hours of treatment, cordycepin alone and cisplatin + cordycepin induced significant expression of phosphorylated p53 (P<0.05, Figure 8B). After 24 hours of treatment, cordycepin, cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel significantly induced expression of phosphorylated p53 in MA-10 cells (P<0.05, Figure 8B). These results indicate that cordycepin, but not paclitaxel or cisplatin, could activate the p53 pathway to induce apoptosis of MA-10 cells.
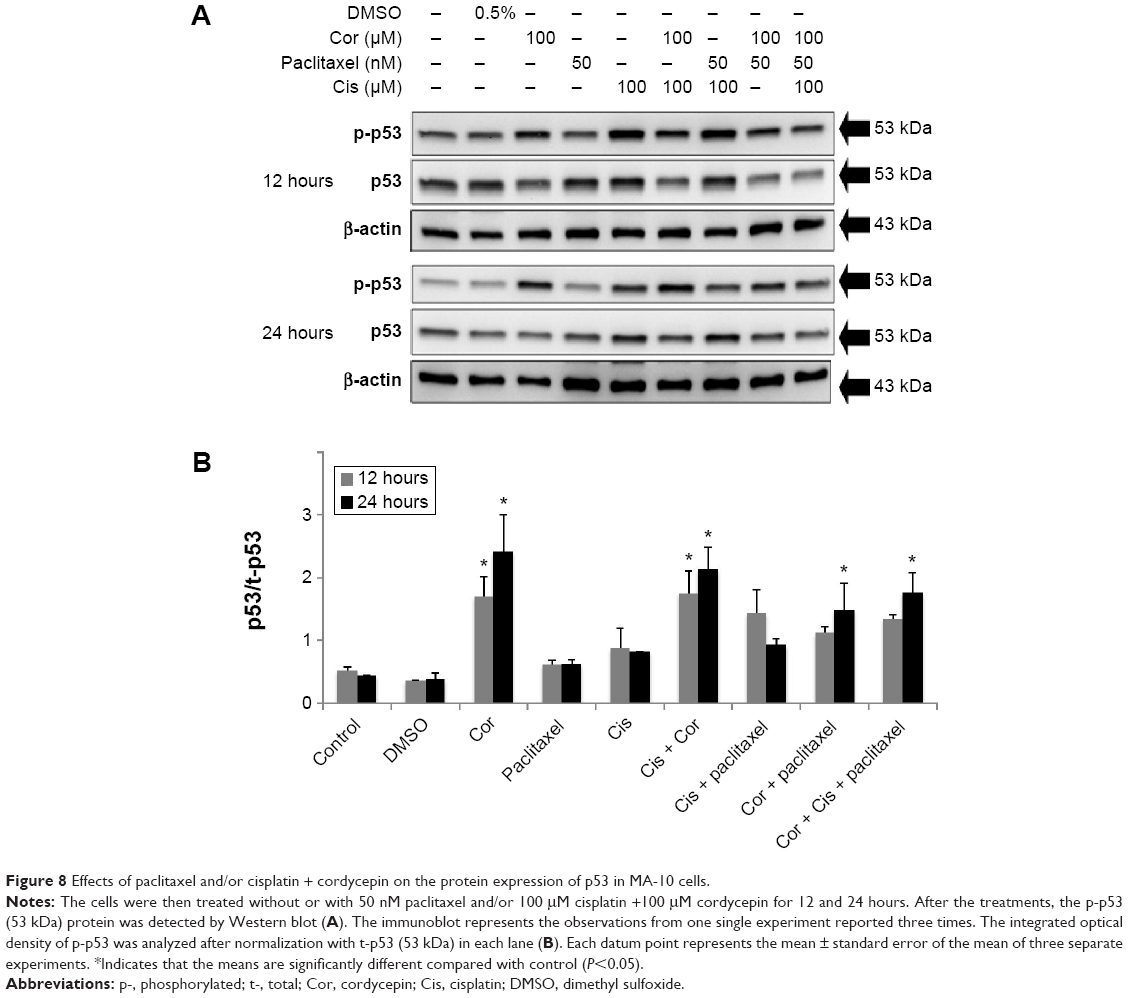 | Figure 8 Effects of paclitaxel and/or cisplatin + cordycepin on the protein expression of p53 in MA-10 cells. |
Discussion
Studies have demonstrated that anticancer agents can have side effects and drug resistance,36–38 and the tendency of cancer treatment in the combination of different chemicals could be more therapeutic to treat cancer. Therefore, we attempted to determine whether the combination of different anticancer agents would have a better effect than each agent used alone in the treatment of testicular cancer.
The results of our present study showed that cordycepin, paclitaxel, and cisplatin-induced blebbing of the MA-10 cell membrane. However, coadministration of cisplatin + cordycepin, cisplatin + paclitaxel, cordycepin + paclitaxel, or cisplatin + cordycepin + paclitaxel induced much more rounding up of MA-10 cells with membrane blebbing. Moreover, we found that coadministration of cisplatin + cordycepin, cisplatin + paclitaxel, cordycepin + paclitaxel, or cisplatin + cordycepin + paclitaxel reduced cell viability to a greater extent than each agent used alone. Based on morphological changes and cell viability studies, the percentage of cell death induced by the combination of cisplatin + cordycepin was approximately 32.42%, which is approximately equal to the sum of the 17% cell death rate achieved by cordycepin 100 μM and the 17% cell death rate achieved by cisplatin 100 μM alone; similarly percentage of cell death induced by the cordycepin + paclitaxel was approximately 28.61%, which was approximately equal to the sum of the 17% cell death rate achieved by cordycepin 100 μM and the 20% cell death rate achieved by paclitaxel 50 nM alone after treatment for 24 hours. We did not observe this effect at 12 hours, but prolonged combination treatment for 24, 36, and 48 hours had a greater antitumor effect in MA-10 cells. Recent studies have demonstrated that combination of different antitumoral treatments can increase the therapeutic efficacy. Sawada et al39 showed that the combination of paclitaxel and cisplatin was more effective in a cisplatin-resistant ovarian carcinoma cell line. Moreover, the additive effect of adriamycin combined with lomustine, prednisolone, cisplatin, VP-16, and Yoshi in inducing death of cultured human lymphoma cells has been demonstrated.40 Thus, our data are consistent with previous reports that drug combinations are more effective than single agents.
Cell cycle arrest could be a major cellular response to DNA damage preceding the decision that a cell should repair itself or die.41 Many of the compounds used as antitumor agents act at multiple steps in the cell cycle, and their effects may be cytostatic or cytotoxic, depending on the status of the cell cycle in the target cells.42 Moreover, studies have shown that paclitaxel,43,44 cisplatin,45,46 and cordycepin30,47 can induce cell cycle arrest at G2/M phase in some cell types, including human bladder cancer, breast cancer, and lung cancer cells, which then proceed to subG1 phase, ending up in apoptosis. According to our results, the proportion of cells arrested at subG1 phase increased in response to treatment with cordycepin or paclitaxel alone and all combination treatments. In contrast, the proportion of cells arrested at G1 phase was significantly reduced. Moreover, the proportion of cells arrested at G2/M phase increased in cells treated with cisplatin + paclitaxel. This phenomenon indicates that a considerable portion of G1 phase cells treated with cisplatin + paclitaxel progressed to G2/M phase. Once cells had progressed into G2/M phase, they became insensitive to the drugs. This phenomenon might coincide with the possible development of resistance after chemotherapy, indicating that cells might change under the pressure of an antitumor agent and even become more proliferative.48 Moreover, other studies indicate that G2/M phase arrest affords cells the opportunity to repair DNA damage before proceeding through mitosis, but this is still unclear.49 Taken together, we observed that cell numbers accumulated in subG1 phase in response to the combination treatments, suggesting that double or triple agent combinations could work more effective to induce DNA fragmentation and apoptosis of MA-10 cells.
It is well known that Fas transduces cell death signaling upon stimulation by Fas ligand, which could activate the caspase pathways.50 Intracellular cell death signaling is mediated by caspase activation at several steps.51 Moreover, other studies have indicated that apoptosis induced by anticancer drugs involves a CD95/CD95 ligand interaction.52 Further evidence have shown that an intrinsic caspase pathway related to the mitochondria could be induced before cell death.53 Therefore, we investigated the mechanism via which caspase is activated by cisplatin, paclitaxel, and/or cordycepin. It has been shown that anticancer agents can activate expression of caspase-8, caspase-9, and caspase-3,54,55 and then cleave PARP,56 which would further obstruct DNA repair and result in apoptosis. In the present study, combination treatments induced more protein expression with additive effect of caspase-8, caspase-9, and caspase-3 and PARP cleavage compared with treatment with each agent alone. Indeed, all treatments, both single and combined, had a better effect at 12 hours with regard to caspase-8, caspase-9, and PARP cleavage, and at 12 and 24 hours for cleavage of caspase-3. It should be noted that cordycepin alone and in combination with cisplatin and/or paclitaxel had a stronger effect in terms of activating cleavage of caspase-8, caspase-9, and caspase-3 and PARP in MA-10 cells when compared with treatment using cisplatin and/or paclitaxel.
The Ras-MAPK pathway is important for orchestrating the response of the cell to external and internal stimuli.23 This pathway is commonly dysregulated in cancers.57 Moreover, many studies have demonstrated that activation of MAPK can stimulate expression of caspase-3 and cleavage of PARP as well as decrease expression of Bcl-2, which mediates the cellular steps in apoptosis of some types of tumor cell.58,59 In the present study, combined treatment for 12 hours with cisplatin + cordycepin, cordycepin + paclitaxel, or cisplatin + cordycepin + paclitaxel induced significantly more expression of phosphorylated ERK when compared with treatment using cisplatin, paclitaxel, or cordycepin alone. Studies have indicated that combination of several antitumor drugs, such as cisplatin, vinblastine, bleomycin, tunicamycin, and actinomycin D, could have additive and/or synergistic effects in testicular cancer through different cellular mechanisms.60 In the present study, we did find remarkable additive effects in MA-10 cells. Interestingly, our studies did show induction of JNK, ERK, and p38 protein phosphorylation. Further, SP600125, PD98059, and PD98059 could reverse the activation of JNK, ERK, and p38 in the combination treatment of cordycepin plus cisplatin and/or taxol, respectively. Thus, our observations are comparable with those of other studies, ie, the combination of different drugs could activate caspase and MAPK pathways to induce apoptosis in various tumor cells.61
Previous studies indicated that the tumor suppressor protein, p53, exerts its inhibitory effect on cell growth by activating and interacting with diverse signaling pathways.34 As a downstream target, p53 protein is phosphorylated and activated by a number of protein kinases in response to stressful stimuli.34 Activation of p53 leads to many outcomes in cells, including cell cycle arrest and apoptosis. It has become clear that the p53 protein can functionally interact with the MAPK pathways. Moreover, both p53 and MAPK signaling pathways are altered in the majority of human tumors.34 Thus, p53 is an important protein that we need to examine and understand. In the present study, cordycepin, cisplatin + cordycepin, cordycepin + paclitaxel, and cordycepin + cisplatin + paclitaxel induced significant expression of phosphorylated p53 in MA-10 cells, implying that activation of p53 protein is important in this event. Indeed, based on the data, cordycepin is an important component in combination treatment for inducing p53 phosphorylation, since cisplatin and paclitaxel alone did not have this effect.
Conclusion
Cordycepin + paclitaxel and/or cisplatin have the ability to induce apoptosis of MA-10 cells, and showed an additive effect when used in combination. Specifically, the combined treatments induced both the intrinsic and extrinsic caspase pathways. Further, the MAPK and p53 signal pathways were also activated. These results strongly suggest that the combination of cordycepin + paclitaxel and/or cisplatin might be more effective than when these agents are used as single agents for chemotherapy in testicular cancer.
Acknowledgments
This work was supported by a grant (CMNCKU10317) from Chi Mei-NCKU Hospital (to FCK and BMH) and a grant (NSC101-2320-B-006-005-MY3) from the National Science Council (to BMH).
Disclosure
The authors report no conflicts of interest in this work.
References
Hiipakka RA, Liao S. Molecular mechanism of androgen action. Trends Endocrinol Metab. 1998;9(8):317–324. | ||
Balasch J, Fábregues F, Casamitjana R, Peñarrubia J, Vanrell JA. A pharmacokinetic and endocrine comparison of recombinant follicle-stimulating hormone and human menopausal gonadotrophin in polycystic ovary syndrome. Reprod Biomed Online. 2003;6(3):296–301. | ||
Jemal A, Siegel R, Ward E, et al. Cancer statistics. CA Cancer J Clin. 2007;57(1):43–66. | ||
Miyata Y, Nomata K, Ohba K, et al. Use of low-dose combined therapy with gemcitabine and paclitaxel for advanced urothelial cancer patients with resistance to cisplatin-containing therapy: a retrospective analysis. Cancer Chemother Pharmacol. 2012;70(3):451–459. | ||
Li QQ, Wang G, Reed E, Huang L, Cuff CF. Evaluation of cisplatin in combination with b-elemene as a regimen for prostate cancer chemotherapy. Basic Clin Pharmacol Toxicol. 2010;107(5):868–876. | ||
Yeruva L, Hall C, Elegbede JA, Carper SW. Perillyl alcohol and methyl jasmonate sensitize cancer cells to cisplatin. Anti-Cancer Drugs. 2010;21(1):1–9. | ||
Kim YH, Shin SW, Kim BS, et al. Paclitaxel, 5-fluorouracil, and cisplatin combination chemotherapy for the treatment of advanced gastric carcinoma. Cancer. 1999;85(2):295–301. | ||
Cunningham KG, Hutchinson SA, Manson W, Hutchinson SA. Cordycepin, a metabolic product isolated from cultures of Cordyceps militaris. Nature. 1950;166(4231):949. | ||
Jen CY, Lin CY, Huang BM, Leu SF. Cordycepin induced MA-10 mouse Leydig tumor cell apoptosis through caspase-9 pathway. Evid Based Complement Alternat Med. 2011;2011:984537. | ||
Pan BS, Lin CY, Huang BM. The effect of cordycepin on steroidogenesis and apoptosis in MA-10 mouse Leydig tumor cells. Evid Based Complement Alternat Med. 2011;2011:750468. | ||
Wu WC, Hsiao JR, Lian YY, Lin CY, Huang BM. The apoptotic effect of cordycepin on human OEC-M1 oral cancer cell line. Cancer Chemother Pharmacol. 2007;60(1):103–111. | ||
Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumor effect of cordycepin (3-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res. 2006;26(1A):43–47. | ||
Thomadaki H, Tsiapalis CM, Scorilas A. Polyadenylate polymerase modulations in human epithelioid cervix and breast cancer cell lines, treated with etoposide or cordycepin, follow cell cycle rather than apoptosis induction. Biol Chem. 2005;386(5):471–480. | ||
Ahn HJ, Kim YS, Kim JU, Han SM, Shin JW, Yang HO. Mechanism of taxol-induced apoptosis in human SKOV3 ovarian carcinoma cells. J Biol Chem. 2004;91(5):1043–1052. | ||
Rowinsky EK, Donehower RC. Paclitaxel (Taxol). N Engl J Med. 1995;332(15):1004–1014. | ||
Rosenberg B. Platinum complexes for the treatment of cancer: why the search goes on. In: Lippert B, editor. Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Zürich, Switzerland: Verlag Helvetica Chimica Acta; 2006. | ||
Johnstone TC, Park GY, Lippard SJ. Understanding and improving platinum anticancer drugs – phenanthriplatin. Anticancer Res. 2014;34(1):471–476. | ||
Fuertes MA, Castilla J, Alonso C, Pérez JM. Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr Med Chem. 2003;10(3):257–266. | ||
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. | ||
Gupta S. Molecular steps of cell suicide: an insight into immune senescence. J Clin Immunol. 2000;20(4):229–240. | ||
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. | ||
Boulares AH, Yakovlev AG, Ivanova V, et al. Role of poly (ADP-ribose) polymerase (PARP) cleavage in apoptosis-caspase 3 resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274(33):22932–22940. | ||
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–2349. | ||
Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28(3):128–136. | ||
Chen YH, Wang JY, Pan BS, et al. Cordycepin enhances cisplatin apoptotic effect through caspase/MAPK pathways in human head and neck tumor cells. Onco Targets Ther. 2013;6:983–998. | ||
Chen YH, Hao LJ, Hung CP, Chen JW, Leu SF, Huang BM. Apoptotic effect of cisplatin and cordycepin on OC3 human oral cancer cells. Chin J Integr Med. 2014;20(8):624–632. | ||
Gupta S. Molecular steps of death receptor and mitochondrial pathways of apoptosis. Life Sci. 2001;69(23):2957–2964. | ||
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. | ||
Chen YH, Leu SF, Jen CY, Huang BM. Effects of sesamol on apoptosis and steroidogenesis in MA-10 mouse Leydig tumor cells. J Agric Food Chem. 2011;59(18):9885–9891. | ||
Lee SJ, Kim SK, Choi WS, Kim WJ, Moon SK. Cordycepin causes p21WAF1-mediated G2/M cell-cycle arrest by regulating c-Jun N-terminal kinase activation in human bladder cancer cells. Arch Biochem Biophys. 2009;490(2):103–109. | ||
Li W, Melton DW. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene. 2012;31(19):2412–2422. | ||
Seidman R, Gitelman I, Sagi O, Horwitz SB, Wolfson M. The role of ERK 1/2 and p38 MAP-kinase pathways in Taxol-induced apoptosis in human ovarian carcinoma cells. Exp Cell Res. 2001;268(1):84–92. | ||
Heinrichs S, Deppert W. Apoptosis or growth arrest: modulation of the cellular response to p53 by proliferative signals. Oncogene. 2003;22(4):555–571. | ||
Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3(12):156–161. | ||
Damia G, Filiberti L, Vikhanskaya F, et al. Cisplatinum and taxol induce different patterns of p53 phosphorylation. Neoplasia. 2001;3(1):10–16. | ||
Monsuez JJ, Charniot JC, Vignat N, Artigou JY. Cardiac side-effects of cancer chemotherapy. Int J Cardiol. 2010;144(1):3–15. | ||
Cavaletti G. Peripheral neurotoxicity of platinum-based chemotherapy. Nat Rev Cancer. 2008;8(1):1. | ||
Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–2094. | ||
Sawada S, Mese H, Sasaki A, Yoshioka N, Matsumura T. Combination chemotherapy of paclitaxel and cisplatin induces apoptosis with Bcl-2 phosphorylation in a cisplatin-resistant human epidermoid carcinoma cell line. Cancer Chemother Pharmacol. 2003;51(6):505–511. | ||
Drewinko B, Green C, Loo TL. Combination chemotherapy in vitro with cis-dichlorodiammineplatinum(II). Cancer Treat Rep. 1976;60(11):1619–1625. | ||
Higginbottom K, Jahnke U, Newland AC, Cotter FE, Allen PD. New alternative phosphorylation sites on the cyclin dependent kinase 1/cyclin a complex in p53-deficient human cells treated with etoposide: possible association with etoposide-induced apoptosis. Apoptosis. 2007;12(10):1847–1855. | ||
Shapiro GI, Harper JW. Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest. 1999;104(12):1645–1653. | ||
Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem. 2004;279(15):15196–15203. | ||
Bacus SS, Gudkov AV, Lowe M, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20(2):147–155. | ||
Sorenson CM, Barry MA, Eastman A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst. 1990;82(9):749–755. | ||
Weir NM, Selvendiran K, Kutala VK, et al. Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Ther. 2007;6(2):178–184. | ||
Palayoor ST, Macklis RM, Bump EA, Coleman CN. Modulation of radiation-induced apoptosis and G2/M block in murine T-lymphoma cells. Radiat Res. 1995;141(3):235–243. | ||
Boulikas T, Vougiouka M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Onco Rep. 2004;11(3):559–595. | ||
Kaufmann SH. Cell death induced by topoisomerase-targeted drugs: more questions than answers. Biochim Biophys Acta. 1998;1400(1–3):195–121. | ||
Nagata S. Fas-mediated apoptosis. Adv Exp Med Biol. 1996;406:119–124. | ||
Suzuki A, Shiraki K. Tumor cell “dead or alive”: caspase and survivin regulate cell death, cell cycle and cell survival. Histol Histopathol. 2001;16(2):583–593. | ||
Wesselborg S, Engels IH, Rossmann E, Los M, Schulze-Osthoff K. Anticancer drugs induce caspase-8/FLICE activation and apoptosis in the absence of CD95 receptor/ligand interaction. Blood. 1999;93(9):3053–3063. | ||
Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18(1):44–51. | ||
Ferrari D, Stepczynska A, Los M, Wesselborg S, Schulze-Osthoff K. Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95- and anticancer drug-induced apoptosis. J Exp Med. 1998;188(5):979–984. | ||
Fulda S, Debatin KM. Sensitization for anticancer drug-induced apoptosis by betulinic acid. Neoplasia. 2005;7(2):162–170. | ||
Wang GQ, Gastman BR, Wieckowski E, et al. A role for mitochondrial Bak in apoptotic response to anticancer drugs. J Biol Chem. 2001;276(36):34307–34317. | ||
Dangle PP, Zaharieva B, Jia H, Pohar KS. Ras-MAPK pathway as a therapeutic target in cancer – emphasis on bladder cancer. Recent Pat Anticancer Drug Discov. 2009;4(2):125–136. | ||
Toh WH, Siddique MM, Boominathan L, Lin KW, Sabapathy K. C-Jun regulates the stability and activity of the p53 homologue. J Biol Chem. 2004;279(43):44713–44722. | ||
Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24(8):1361–1367. | ||
Einhorn LH, Donohue JP. Improved chemotherapy in disseminated testicular cancer. J Urol. 1977;117(1):65–69. | ||
Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98(9):2603–2614. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.