")
Back to Journals » OncoTargets and Therapy » Volume 7
Angiogenesis and antiangiogenic agents in cervical cancer
Authors Tomao F , Papa A, Rossi L, Zaccarelli E, Caruso D, Zoratto F, Benedetti Panici P, Tomao S
Received 23 May 2014
Accepted for publication 16 August 2014
Published 3 December 2014 Volume 2014:7 Pages 2237—2248
DOI https://doi.org/10.2147/OTT.S68286
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Federica Tomao,1 Anselmo Papa,2 Luigi Rossi,2 Eleonora Zaccarelli,2 Davide Caruso,2 Federica Zoratto,2 Pierluigi Benedetti Panici,1 Silverio Tomao2
1Department of Gynecology and Obstetrics, Sapienza University of Rome, Policlinico Umberto I, Rome, 2Department of Medico-Surgical Sciences and Biotechnologies, Sapienza University of Rome, Oncology Unit, ICOT, Latina, Italy
Abstract: Standard treatment of cervical cancer (CC) consists of surgery in the early stages and of chemoradiation in locally advanced disease. Metastatic CC has a poor prognosis and is usually treated with palliative platinum-based chemotherapy. Current chemotherapeutic regimens are associated with significant adverse effects and only limited activity, making identification of active and tolerable novel targeted agents a high priority. Angiogenesis is a complex process that plays a crucial role in the development of many types of cancer. The dominant role of angiogenesis in CC seems to be directly related to human papillomavirus-related inhibition of p53 and stabilization of hypoxia-inducible factor-1α. Both of these mechanisms are able to increase expression of vascular endothelial growth factor (VEGF). Activation of VEGF promotes endothelial cell proliferation and migration, favoring formation of new blood vessels and increasing permeability of existing blood vessels. Since bevacizumab, a recombinant humanized monoclonal antibody binding to all isoforms of VEGF, has been demonstrated to significantly improve survival in gynecologic cancer, some recent clinical research has explored the possibility of using novel therapies directed toward inhibition of angiogenesis in CC too. Here we review the main results from studies concerning the use of antiangiogenic drugs that are being investigated for the treatment of CC.
Keywords: cervical cancer, angiogenesis, human papillomavirus, bevacizumab, target therapies
Introduction
In the USA, there will be an estimated 12,360 new cases of cervical cancer (CC) in 2013, with 4,020 cancer-related deaths, making CC the twelfth most common cancer in women and the second cause of death in women aged 20–39 years.1–3
The leading cause of CC is latent infection by human papillomavirus, in particular subtypes 16 and 18. Its pathogenic action is related to E6 and E7 proteins: E6 promotes the degradation of p53 while E7 inactivates retinoblastoma protein.4 Degradation of p53 could be responsible of activation of angiogenesis through production of vascular endothelial growth factor (VEGF)5–8 and downregulation of a potent angiogenesis inhibitor, thrombospondin-1.9,10 Moreover, in recent years, there has been increasing interest in E5 protein, which seems to be involved in activation of epidermal growth factor receptor, in the modulation of the inflammation process, and in induction of angiogenesis through VEGF.11
The angiogenesis process, defined in 1971 by Folkman as a critical point for the growth of tumors, is controlled at different levels. In particular, the transition from the avascular to the vascular phase is termed the “angiogenic switch” of the tumor and is thought to be a key element in the clinical observation of tumor dormancy.12 Additionally, Folkman proposed that neovascularization is a vital process in metastatic spread by allowing malignant cells to enter into the circulation.13,14
Regarding the treatment of CC, 30 years after the introduction of cisplatin, little improvement have been made with the introduction of new drugs and combinations; in fact, at the present time, combination chemotherapy does not show a long-term clinical benefit, and in advanced disease, the overall survival (OS) does not reach 1 year.15–18 In this sense, use of novel therapeutic regimens with the association of targeted agents could be useful to counteract this situation. The aim of this review was to analyze the clinical activity and safety profiles of antiangiogenic drugs that have been investigated for the treatment of CC.
Angiogenesis and cervical cancer
Angiogenesis occurs through a dynamic balance of proangiogenic and antiangiogenic factors favoring physiological homeostasis. In normal tissue, the vasculature remains quiescent (Figure 1), but in neoplastic tissues, upregulation of proangiogenic factors, eg, VEGF, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and angiopoietins, and downregulation of antiangiogenic factors, eg, thrombospondin, angiostatin, and endostatin, tips the balance in favor of the “angiogenic switch”, with the occurrence of neovascularization.19 Many tissue environmental factors, including hypoxia and low pH, hormones (eg, progesterone, estrogen), growth factors (eg, endothelial growth factor, transforming growth factor-², FGF, PDGF, insulin-like growth factor-1), and cytokines (eg, interleukin-1 and interleukin-6) stimulate VEGF expression. In addition to exogenous factors, many tumorigenic mutations lead to upregulation of VEGF. These can include mutations in cellular oncogenes, such as the src, ras, and bcr-abl, and in tumor suppressor genes too, such as p53, p73, and VHL Lindau.
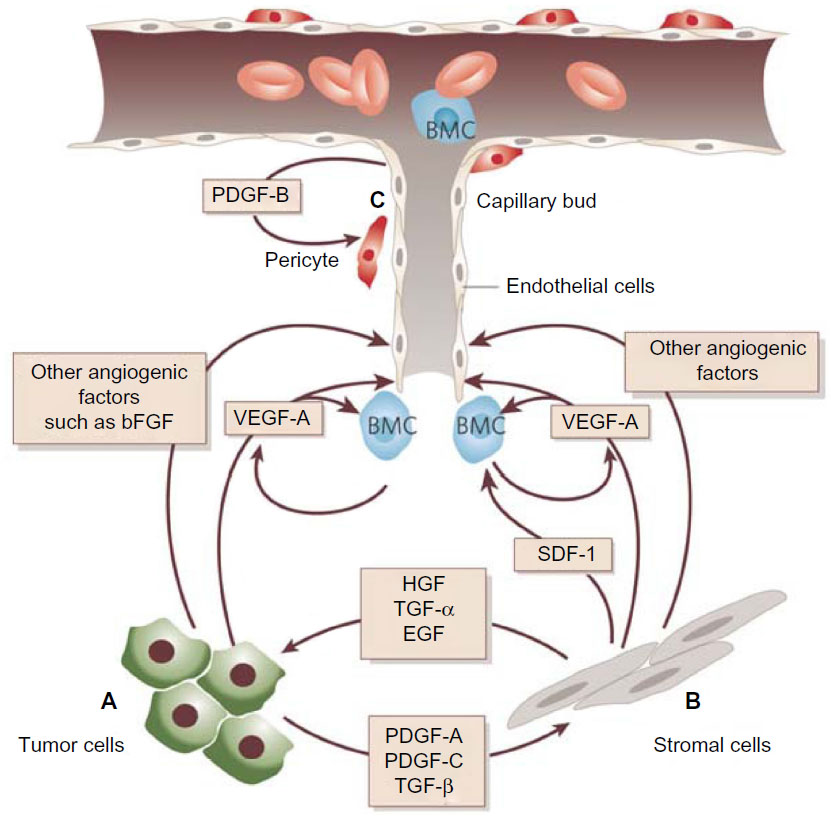 | Figure 1 Tumor angiogenesis. |
The role of FGF signaling in angiogenesis is well established.20,21 FGF-1 and, in particular, FGF-2 are potent proangiogenic factors.22,23 During angiogenesis, FGFs are secreted from the extracellular matrix from tumor cells in an autocrine fashion and from endothelial/stromal cells in a paracrine fashion. FGF-2 also increases the expression of other proangiogenic proteins, including angiopoietin-2 and VEGF.24,25 Moreover, there is full evidence of crosstalk between FGF-2 and VEGF with preclinical data both from in vitro and in vivo systems; moreover, a synergic activity was demonstrated between the two, with differing effects on vessel size and function.25–28
Members of the VEGF family are the most potent proangiogenic factors. VEGF is produced by cancer cells and is related to the invasive and metastatic potential of human tumors.29 Activation of VEGF promotes proliferation and migration of endothelial cells with formation of new blood vessels; moreover, it increases the permeability of existing blood vessels, allowing leakage of multiple plasma proteins, including those playing a crucial role in angiogenesis.30
VEGF is secreted in response to hypoxia, acidosis, and mechanical stress. Exposure of tumor cells to hypoxia leads to production of hypoxia-inducible factor-1α, which forms a complex with hypoxia-inducible factor-1²; this complex is responsible of the production of growth factors like VEGFs.31,32 The VEGF axis consists of a family of structurally related proteins, ie, VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor, VEGF-A being the major mediator of angiogenesis.33 Recently, it has been suggested that VEGF also exerts an immunosuppressive effect in cancer as it was correlated with low levels of interleukin-12, inhibition of maturation of dendritic cells, low numbers of natural killer T-cells, and upregulation of regulatory T-cells.34–37 VEGF also inhibits apoptosis of the newly formed hyperpermeable blood vessels.38 These proteins interact with three VEGF tyrosine kinase receptors (VEGFR1–3) to initiate a cascade of downstream signaling pathways, promoting migration, proliferation, and survival of endothelial cells and leading to formation of new blood vessels. VEGFR1 and VEGFR2 principally regulate angiogenesis, whilst VEGFR3 plays a lesser role in angiogenesis but is critical for lymphangiogenesis, so is implicated in the production of ascites and lymphatic dissemination of metastatic factors.39
Angiopoietins (Angs) are ligands of the endothelial cell receptor Tie-2. Tie-2 belongs to the receptor tyrosine kinase family and is mainly expressed in vascular endothelial cells and early hematopoietic cells, and is involved in the stability of mature vessels and proliferation of endothelial cells.40 In vitro and in vivo experiments showed that Ang-1, Ang-2, and Tie-2 compose a system that can regulate vascular quiescence and endothelial stability and, subsequently, maintain the maturation and stability of vessel networks.41
Ang-1 and Ang-2 have important roles in angiogenesis, but the nature of their contributions is distinct. Ang-2 is mainly secreted by endothelial cells at sites of active vascular remodeling, and is involved in tumor initiation.42 Ang-1 is expressed by mural cells under physiological conditions and plays a key role in maintaining the integrity of existing vessels. The evidence shows that an imbalance between Ang-2/Ang-1 may be attributed to interruption of pre-existing vascular stability; this pathological process plays an important role in early initiation and activation of angiogenesis during malignancy.43
Oliner et al used antibodies to inhibit the interaction between Ang-2 and Tie-2, and their results showed that growth of cancer cells was markedly suppressed and accompanied by a reduction in endothelial cell proliferation.44 Given that Ang-2 promotes the proangiogenic action of VEGF45–47 and VEGF upregulates Ang-2 expression in endothelial cells,48 inhibition of Ang-2 and VEGF together could have complementary actions by reducing sprouting angiogenesis and tumor vascularity.47,49,50
Some early evidence suggests that inhibition of Ang-2 may have additional beneficial effects in cancer therapy, probably mediated by mechanisms that are separate from the destabilizing effect of Ang-2 on tumor vessels. Recent studies have demonstrated that tumor-associated monocytes or macrophages expressing Tie-2 receptors can be recruited and/or activated by angiopoietins; these factors seem to promote tumor angiogenesis by release of some proteases that are able to liberate sequestered VEGF from the tumor matrix.51,52 Further studies will be needed to determine whether inhibition of Ang-2 signaling suppresses a macrophage-dependent mechanism that complements the direct effects of Ang-2 inhibition on endothelial cells.53
The role of angiogenesis in invasive CC has been known from a long time,54 but in recent years some studies have demonstrated that angiogenesis is relevant also in premalignant lesions of the cervix;55 in fact, microvessel density (MVD) increases with grade of cervical intraepithelial neoplasia.56 Evaluation of MVD from 111 biopsies of the cervix and the association with clinical outcome led to the conclusion that high vascularity is associated with worse survival (50% at 5 years), while low vascularity shows better results (65% at 5 years).57 A variety of markers are used to measure MVD, including CD105, von Willebrand factor, CD31, and CD34, but are nonspecific for endothelial cells, so their clinical utility may be limited.58
CD105 (endoglin), a component of transforming growth factor-²,59 is expressed almost exclusively on endothelial cells in solid tumors during neoangiogenesis, and it modulates angiogenesis by regulating cellular proliferation, differentiation, and migration. In CC specimens, CD105-positive vessels have been found, showing the association between this tumor and the angiogenic process; the presence of CD105-positive vessels has shown an association with risk of lymph node metastasis,60,61 worse progression-free survival (PFS), and unfavorable OS.
CD31 has been associated with a prolonged PFS and OS, and shown to be an independent prognostic factor in women with high-risk, early-stage CC treated with radiation therapy or chemoradiation.62 There is good evidence that new CD31-positive vessels result in a more organized endothelium, while CD105-positive vessels show a disorganized endothelial structure.63
Analysis of MVD, measured with CD31, CD105 and Factor 8, used as markers of angiogenesis, yielded controversial results: some studies showed a worse clinical outcome in women with high tumor MVD64 while others showed better survival in those with higher MVD;65,66 however, others found no association between MVD and outcome.67,68
Analysis of VEGF in 50 CC specimens using a monoclonal antibody showed increased VEGF expression in cervical intraepithelial neoplasia grade III and squamous cell carcinoma when compared with controls;69 further, levels of VEGF and MVD showed a strong association. Nevertheless, the increase in tumor MVD has been associated with decreased delivery of chemotherapy and worse clinical outcome in a variety of solid tumors.70–73
The possibility of an association between CC and VEGF gene polymorphisms is fascinating; some of these proteins appear to be linked with higher VEGF expression and some other with a lower expression.74–77 An experience from 199 patients showed that VEGF-2578 A/A and VEGF + 405G>C polymorphisms are associated with a lower and higher risk of CC, respectively. These results are not conclusive because experiences in other cancers showed different results.78
Further, in CC, human papillomavirus plays an important role in angiogenesis through the action of E6 protein. It is responsible for p53 dysregulation and occurs via two mechanisms. The first one is DNA damage that lead to arrest of p53 induction and its ubiquitination, with subsequent degradation of the protein. One of the consequences of the degradation of p53 is upregulation of VEGF.79 The second mechanism is the activation of hypoxia-inducible factor-1α, which increases VEGF production.80
As described, angiogenesis plays a role of primary importance, not only in initiation of cancer of the cervix, but also in proliferation and progression of the disease, adversely affecting the prognosis. Therefore, in recent years, different pathways of angiogenesis have been evaluated as potential therapeutic targets in the treatment of CC.
Antiangiogenic target therapies
Blocking proangiogenic factors has been shown to be an effective strategy for controlling CC tumor growth.
There are two primary strategies to inhibit the VEGF pathway, ie, inhibition of the VEGF ligand with antibodies or soluble receptors and inhibition of the VEGFR with tyrosine kinase inhibitors (TKIs) or receptor antibodies. Therapies that specifically target the VEGF ligand or its receptors, VEGFR-1 and VEGFR-2, inhibit only the VEGF pathway and therefore inhibit angiogenesis without disrupting off-target pathways. In contrast, TKIs targeting the receptor have a wider range of inhibitory effects and may disrupt other secondary pathways that are mediated through receptor kinases. To improve the therapeutic benefits and counteract compensatory escape mechanisms, another approach to antiangiogenic therapy is the simultaneous targeting of multiple angiogenic pathways at once, including the PDGF and FGF pathways.
FGF and its receptors play an important role in the development of resistance to VEGF pathway inhibitors.81 In preclinical models, blocking VEGF, PDGF, and FGF pathways suppresses tumor angiogenesis and MVD to a greater extent than selectively blocking the VEGF pathway.
Bevacizumab in cervical cancer
Bevacizumab is a recombinant humanized monoclonal antibody of the IgG1 isotype that binds to all isoforms of VEGF with high specificity and affinity, resulting in potent VEGF neutralizing activity; thus, this agent is able to inhibit endothelial cell proliferation and vessel formation.82,83 It has been successfully studied in many solid tumors, including colon,84 lung,85 breast,86 kidney, brain,87 and ovarian88,89 cancers, with the result that it was the first clinically available antiangiogenic agent registered and extensively used in the USA.90 These findings validated inhibition of the VEGF signaling pathway as an important treatment target in cancer therapy.
In 2006, a small retrospective study suggested encouraging antitumor activity of bevacizumab in combination with 5-fluorouracil (or capecitabine) in six heavily pretreated patients with recurrent CC. Treatment was well tolerated. Clinical benefit was noted in 67% of the subjects and the median time to progression for the four women who experienced clinical benefit was 4.3 months (Table 1).91
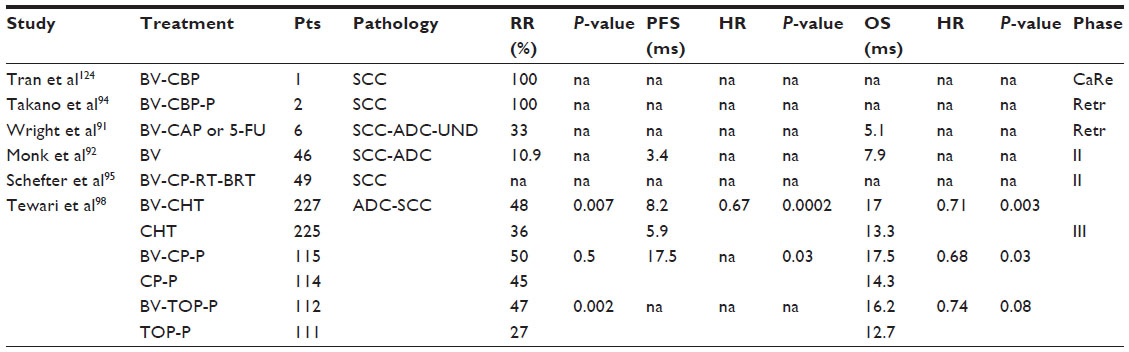 | Table 1 Bevacizumab studies in cervical cancer |
In the protocol for the Phase II Gynecologic Oncology Group (GOG) 227C study, bevacizumab was analyzed in 46 patients with persistent or recurrent squamous cell carcinoma of the cervix. The results suggested that bevacizumab is not only active in treating recurrent CC but also safe (Table 1).92
In another interesting study, 12 cases with recurrent or metastatic CC after intensive treatment including radiotherapy were treated by weekly association of bevacizumab and paclitaxel/carboplatin with or without sorafenib. On evaluation by RECIST (Response Evaluation Criteria in Solid Tumors), 2 of 12 patients had a complete remission and five had a partial remission. Four of seven cases treated with sorafenib showed a partial remission, and three of five patients without sorafenib obtained a partial remission (Table 1).93
Takano et al attempted to explore the effects of weekly bevacizumab and gemcitabine/oxaliplatin with or without dasatinib in ten patients with recurrent or metastatic CC after intensive treatment including radiotherapy. On RECIST evaluation, one patient experienced a complete remission and four experienced partial remission. The overall response rate was 50%, and responses were observed in four of five cases treated with dasatinib and in one of five patients without dasatinib (Table 1). Neurotoxicity, nasal bleeding, and general fatigue were frequently observed, but were all grade 1.94
In 2012, an interesting study evaluated the safety and efficacy of addition of bevacizumab to standard chemoradiotherapy in 49 untreated patients with locally advanced CC (stage IB–IIIB). The median follow-up was 12.4 months. There were 15 protocol-specified treatment-related adverse events within 90 days from the start of treatment, the most common being hematological.95
Another study is evaluating the role of bevacizumab in combination with radiotherapy and cisplatin in patients with previously untreated locally advanced CC.96 Recently, the combination of topotecan, cisplatin, and bevacizumab resulted in an active but highly toxic regimen in patients with recurrent or persistent CC not amenable to treatment with curative intent.97
Based on these encouraging results, between 2009 and 2012 in the USA and Spain, 452 women with stage IVB or persistent and recurrent CC after standard treatment were enrolled in GOG 240, a randomized Phase III trial. This study was designed to answer two important questions, ie, whether topotecan in combination with paclitaxel was superior to the association of cisplatin and paclitaxel and whether the addition of bevacizumab 15 mg/kg to either regimen improved OS. More than 70% of patients in each group had previously received platinum-based therapy as a radiosensitizing strategy. Patients were randomly assigned to one of four treatment groups; two of the treatment groups received bevacizumab. A total of 225 patients received chemotherapy alone and 227 also received bevacizumab. In an analysis conducted in 2012, it was stated that topotecan plus paclitaxel was not superior to the standard therapy of cisplatin plus paclitaxel (P=0.880). The study met its primary endpoint of demonstrating improved OS in patients who received bevacizumab. At a median follow-up of 20.8 months, bevacizumab-treated patients lived for a median of 3.7 months longer than those who did not receive bevacizumab (hazard ratio 0.71, P=0.0035; Table 1).98
The PFS and response rate were also better in patients who received bevacizumab than in those who received chemotherapy alone (Table 1).98 There were 28 complete responses in the patients who received bevacizumab and 14 in the chemotherapy only group. Bevacizumab conferred a significant survival benefit when added to cisplatin-paclitaxel chemotherapy (P=0.0348) but not when added to topotecan-paclitaxel chemotherapy (P=0.08969, Table 1).98 The advantage seen with bevacizumab persisted in some subsets of patients, including those aged 48 and 56 years and those with recurrent/persistent disease, but not in the 76 patients with metastatic disease and those with squamous histology. Most importantly, when the disease was detected in a previously irradiated pelvis, bevacizumab was still effective.
For the first time, a targeted agent significantly improved OS in gynecological cancer. The nearly 4-month increase in median OS with the addition of bevacizumab to chemotherapy in women with recurrent CC is considered to be clinically significant. Nevertheless, there was no crossover, which enables investigators to get the purest survival data. These findings are important because they are likely to change clinical practice and provide an opportunity to improve outcome in patients with recurrent CC, a subset of patients who in the past had very limited treatment options.
However, patients receiving bevacizumab experienced more side effects than those who did not. These side effects were consistent with those previously known to be associated with bevacizumab, and included hypertension, neutropenia, and thromboembolism, or formation of blood clots. Specifically, treatment with bevacizumab was associated with more grade 3–4 bleeding, thrombosis/embolism, and gastrointestinal fistula. Although both thromboembolic events and fistula were increased in the bevacizumab arms, these rates were relatively low, being below 10%.
Quality of life was investigated using the FACT-Cx TOI (Functional Assessment of Cancer Therapy–Cervix Trial Outcome Index scale) which assesses physical and functional well-being specific to CC, where a between-group overall score difference of 5 or more would indicate a clinically significant detrimental impact on quality of life.98 In this study, the FACT-Cx score difference between patients receiving chemotherapy alone and those aged 48 and 56 years receiving bevacizumab was only 1.2 points, indicating that the increased survival with bevacizumab did not occur at a cost of decreased quality of life.98
There are some open questions about the role of bevacizumab in CC. Only about one-fifth (17%) of patients had metastatic disease, so the benefits of bevacizumab are uncertain in this population; nevertheless, in our opinion, it is very important to evaluate the activity of bevacizumab in studies investigating the role of the drug in earlier disease stages. The data suggest that bevacizumab may have less benefit in patients with adenocarcinoma than in those with squamous cancer, suggesting a need to discover predictive biomarkers of response. Extending the global reach of antiangiogenic therapies for advanced CC is critical because the countries with the highest incidence and mortality rates for this disease also have the lowest annual health care spending.98
Regarding the possibility of different efficacy of bevacizumab in relation to the histological type of CC, there are insufficient data to make a definitive judgment regarding adenocarcinoma histology because the numbers of patients with this histology are inadequate in the studies exploring bevacizumab in CC as reported in the literature; in a study by Tewari et al, bevacizumab seemed to be less effective in the subgroup of patients with adenocarcinoma histology (n=86).98
We still do not know the precise mechanisms that cause resistance to bevacizumab. ML18147 and BRITE were the first two randomized Phase III studies demonstrating that OS and PFS were significantly prolonged with continued bevacizumab plus chemotherapy compared with chemotherapy alone as second-line treatment in patients with metastatic colorectal cancer progressing after a first-line bevacizumab-containing regimen. These data indicate that the antiangiogenic effects of bevacizumab might persist despite the development of resistance to first-line chemotherapy.
Consistent with these results, the use of bevacizumab beyond progression might be of great interest in order to investigate a novel therapeutic approach in CC. Eventually, it could be interesting to associate bevacizumab with pemetrexed, considering that it showed moderate activity against advanced/recurrent CC and safety in association with bevacizumab in the treatment of ovarian cancer.
Sunitinib
Sunitinib malate is an oral drug that inhibits cell proliferation and angiogenesis. It is a multitargeted TKI of VEGFR-1, VEGF-2, VEGF-3, PDGFR-α, PDGFR-², c-KIT (the stem cell factor receptor), and FLT3.99–102 It has been approved for renal cancer and imatinib-resistant gastrointestinal stromal tumors. In a Phase II study of sunitinib in pretreated patients with locally advanced or metastatic CC, no objective responses were observed (Table 2). A higher rate of fistula formation (26%) was detected when compared with what has been observed in Phase II studies of other biologics.103
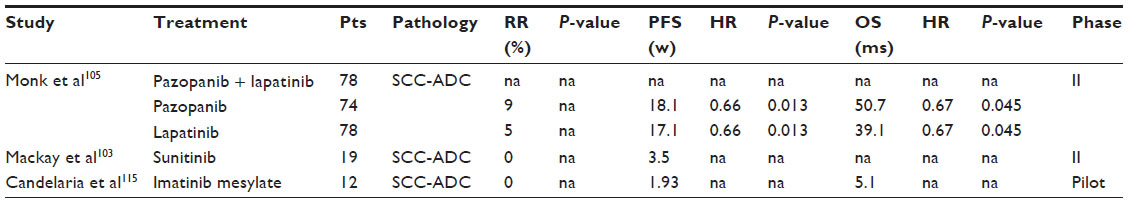 | Table 2 Antiangiogenetic therapies in cervical cancer |
Pazopanib
Pazopanib is an orally administered, multitargeted receptor TKI of VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-α, PDGFR-², and c-kit. It blocks tumor growth and inhibits angiogenesis, and is approved for renal cancer.104 A randomized Phase II study has been conducted comparing pazopanib, lapatinib, or a combination of both in patients with previously treated advanced CC. This study demonstrated the benefit of pazopanib, based on the prolonged PFS and favorable toxicity profile (Table 2).105 Six patients (8%) in the pazopanib arm and three patients (4%) in the lapatinib arm achieved a confirmed partial remission. The most commonly observed toxicities with pazopanib were diarrhea, nausea, hypertension, and anorexia. Only three patients developed fistula, which led to discontinuation of treatment. An updated analysis of this study has revealed no differences in OS between the two arms (Table 2).106
Brivanib
Brivanib alaninate is an orally available small molecule that inhibits in a selective manner both VEFGR and the fibroblast growth factor receptor (FGFR).107 A crosstalk between FGFR and VEGFR in angiogenesis has been demonstrated, with some data pointing at potential mechanisms of resistance to VEGFR inhibitors being actually mediated by the FGFR axis.20,108,109
Brivanib belongs to the first-generation FGFR inhibitors,110 so its primary target is the VEGFR. It exerts antiangiogenic and antitumor effects by inhibition of VEGFR. FGFR also plays an important role in promoting basic FGF-mediated angiogenesis and tumor cell proliferation, and can function as a compensatory signal for VEGFR. Therefore, suppression of FGFR by this first-generation inhibitor could attenuate the compensation of FGFR for VEGFR, thereby providing more pronounced antitumor effects. Although there are several first-generation FGFR inhibitors in clinical trials, their primary target is not FGFRs, and they have only a supporting role in cancer treatment, which precludes our efforts in better understanding how FGFR may affect clinical efficacy.111
The preclinical activity of brivanib has been evaluated in xenografts, showing dose-dependent tumor growth inhibition in breast (H3396), colon (HCT/VM46), and lung (L2987) models.112 Brivanib alaninate has shown potent antitumor activity when dosed orally on a once-daily schedule, with complete induction of tumor stasis. With the termination of dosing, tumor xenografts resume growth.113 Consequently, the anticipated effects of this therapeutic approach predict tumor stasis rather than regression; therefore, antiangiogenic agents are expected to be used either in conjunction with standard therapy or in an adjuvant setting. The Phase II GOG study evaluating the activity of brivanib as a single agent in the second-line treatment of persistent or recurrent CC has now stopped participant recruitment.114
Imatinib
Imatinib is a 2-phenylaminopyrimidine derivative that functions as a specific inhibitor for the tyrosine kinase domain in abl (the Abelson proto-oncogene), c-kit, and PDGFR. It has demonstrated remarkable clinical efficacy in patients with chronic myeloid leukemia and malignant gastrointestinal stromal tumors. Candelaria et al presented a pilot study evaluating imatinib mesylate as second-line treatment for recurrent or metastatic CC expressing PDGFR-α. All patients expressed PDGFR-α in more than 10% of malignant cells, whereas only four coexpressed PDGFR-². No objective responses were observed (Table 2).115
Novel targeted agents with antiangiogenic properties
Unfortunately, antiangiogenic effects in clinical oncology are transient, due to the development of drug resistance.116 For this reason, the study of other antiangiogenic factors may result in more clinically meaningful responses.
A preclinical study has shown that inhibition of Ang-1 expression in human CC HeLa cells greatly reduces xenografted tumor growth in mice as a result of decreased tumor angiogenesis and increased tumor cell apoptosis. Moreover, increased Ang-1 expression promotes in vivo growth of human CC by promoting tumor angiogenesis and increasing tumor vessel plasticity.117 These data would support targeting the Ang-Tie2 system for therapeutic intervention in CC. There are two Ang traps (which function by sequestering Angs) in early clinical development: AMG386 (trebananib) and PF 4856884 (also known as CVX-060).40
AMG386 is a peptide-Fc fusion protein targeting angiogenesis by inhibiting binding of both Ang-1 and Ang-2 to the Tie-2 receptor, thereby inhibiting vascular maturation and reducing the impact of VEGF stimulation.117 In a randomized Phase II trial, the addition of AMG386 to low-dose weekly paclitaxel for recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, demonstrated prolongation of PFS from 4.6 to 7.2 months with paclitaxel-AMG386 (hazard ratio 0.76; P=0.165). More than 50% of the 161 patients had platinum-resistant disease. Grade 3 adverse events occurring more frequently were hypokalemia, peripheral edema, and hypertension.118,119
Recently, Monk et al presented the results of a randomized, double-blind, Phase III trial comparing paclitaxel plus trebananib with paclitaxel and placebo in patients with recurrent epithelial ovarian cancer. Patient eligibility criteria included women previously treated with three or fewer regimens and a platinum-free interval of less than 12 months. The results showed a statistically significant difference in PFS (7.4 months in the trebananib arm versus 5.4 months in the control arm, hazard ratio 0.66, P<0.001) and a 34% reduction in the risk of disease progression or death. The response rate increased from 30% with placebo to 38% with trebananib, and the interim OS did not show any significant difference between groups.120
In a randomized, double-blind, placebo-controlled Phase II study, administration of trebananib plus FOLFIRI did not prolong PFS compared with placebo plus FOLFIRI in patients with previously treated metastatic colorectal cancer; in this experience, there was a trend toward an improved objective response rate. Although trebananib plus FOLFIRI did not improve PFS in this study, the evidence continues to support the concept of antiangiogenesis as a treatment approach in second line with FOLFIRI schedule, including patients who have previously received angiogenesis inhibitors.121 It is possible that treatment approaches incorporating inhibitors of the angiopoietin-Tie2 axis could have a role if administered at different doses/schedules in less advanced disease and/or if administered in combination with other targeted agents (eg, VEGF inhibitors).122
Currently, trebananib plus bevacizumab is being evaluated in a Phase II study as first-line therapy in patients with mCRC.123 Therefore, great interest is directed toward future studies that should confirm the potential antitumor activity of this target therapy in metastatic CC.
Conclusion
Women with metastatic or recurrent CC have very few therapeutic options. While surgery (in early-stage disease) or chemoradiation therapy (in later-stage disease) can be effective, there are very limited treatment options for patients with advanced CC and for those whose cancer relapses after initial treatment; moreover, drug resistance is a very important clinical problem in this disease. Unlike some other solid cancers, CC does not really respond to different chemotherapies. In fact, the standard regimens probably extend survival in few patients.
The majority of women with recurrent CC have received primary treatment with cisplatin-based chemoradiation for locally advanced disease. With widespread adoption of chemoradiation protocols, platin-based therapies would be less effective at recurrence due to acquired drug resistance. In 2009, the Phase III GOG 204 trial explored the activity of four platinum-based chemotherapy doublets in recurrent CC, closing the study for futility; it was discovered that many of the women had platinum-resistant disease because they had received platinum as part of their prior chemoradiation therapy.
Recently, for the first time, a targeted agent has demonstrated an important improvement in gynecologic cancer. In the planned interim analysis of the GOG 240 study, bevacizumab significantly improved OS when added to chemotherapy for patients with recurrent/persistent or metastatic CC. The clinical rationale for including bevacizumab in this study was that VEGF inhibition had shown benefit in a number of solid tumors and angiogenesis is a very active process in CC, known to be a hallmark of invasive disease. Moreover, targeting VEGF in the setting of CC is an extraordinarily good rationale, based on the pathophysiological role of human papillomavirus infection.
More than 70% of patients in both groups had received prior platinum-based therapy; the groups were well matched with regard to age, histology, race, disease stage, and performance status. The initial interim analysis showed that the topotecan-paclitaxel regimen was neither superior nor inferior to the cisplatin-paclitaxel regimen, while the second interim analysis did show superiority when bevacizumab was added to either chemotherapy regimen. The survival analysis conducted earlier in 2013 led the American Society of Clinical Oncology to make a rare exception and allow the abstract into the public domain well in advance of its presentation.
The improvement is felt to be clinically meaningful in a population of patients that does not respond to chemotherapy very well. Moreover, there was no crossover, which enabled the investigators to get the purest survival data. The results may not be applicable to metastatic patients, given that only 17% of those in this trial had metastatic disease. In this study, the FACT-Cx score difference between patients receiving chemotherapy alone and those receiving chemotherapy plus bevacizumab was only 1.2 points, indicating that the increased survival with bevacizumab did not come at a cost of decreased quality of life.
Moving forward, based on the GOG 240 results, the incorporation of anti-VEGF therapy into primary treatment for locally advanced disease should be considered. Furthermore, as already proved in the treatment of colorectal cancer the use of bevacizumab beyond progression might be of great interest in order to investigate a novel therapeutic approach in CC.
These data open up the opportunity to study other classes of antiangiogenic agents, including both VEGF-dependent molecules and non-VEGF-dependent molecules. The use of TKI is based on their multitarget mechanisms but they have not shown the expected benefits, but only a major incidence of adverse events. However, we can make some considerations about the role of TKIs in the treatment of CC. In the Phase III bevacizumab study, only 17% of enrolled patients had metastatic disease, in the Phase II study of pazopanib there were only six patients with metastatic disease, and in the Phase II study of sunitinib this number is unknown. The different efficacy results with bevacizumab, with respect to sunitinib and pazopanib, could suggest that the ultimate setting to evaluate the best benefit from these TKIs is not in early or locally advanced disease but in metastatic disease. Moreover, in the study by Tewari et al, bevacizumab was given as first-line treatment, whereas sunitinib and pazopanib were tested in patients already heavily treated with chemotherapy, this difference could suggest that the role of TKI would be experienced in early line of treatment.
GOG is evaluating the activity of brivanib in the second-line treatment of persistent or recurrent CC (ClinicalTrials.gov NCT01267253); this agent could attenuate the compensation of FGFR for VEGFR, thereby providing more pronounced antitumor effects respect to anti-VEGF. Regarding imatinib, despite its lack of activity as a single-agent, further studies in CC are justified to better define the status of imatinib targets in this tumor and to investigate its activity in combination with cytotoxic drugs. Although AMG386 with chemotherapy did not improve PFS in colon cancer, we can hypothesize that it could be useful in CC because it has shown positive results in the treatment of ovarian cancer.
The discovery of survival gains, such as those conferred by anti-VEGF therapy, need to be followed by cost-effectiveness studies to better reevaluate how to balance the social burden and, at the same time, provide these therapies to those who are in the greatest need.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. | |
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. | |
Howlander N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2009. Bethesda, MD, USA: National Cancer Institute; 2012. | |
Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98:1505–1511. | |
Lopez-Ocejo O, Viloria-Petit A, Bequet-Romero M, Mukhopadhyay D, Rak J, Kerbel RS. Oncogenes and tumor angiogenesis: the HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene. 2000;19: 4611–4620. | |
Lee JS, Kim HS, Jung JJ, Lee MC, Park CS. Expression of vascular endothelial growth factor in adenocarcinomas of the uterine cervix and its relation to angiogenesis and p53 and c-erbB-2 protein expression. Gynecol Oncol. 2002;85:469–475. | |
Lee JS, Kim HS, Park JT, Kim YB, Lee MC, Park CS. Expression of vascular endothelial growth factor in the progression of cervical neoplasia and its relation to angiogenesis and p53 status. Anal Quant Cytol Histol. 2003;25:303–311. | |
Toussaint-Smith E, Donner DB, Roman A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene. 2004;23:2988–2995. | |
Wu MP, Tzeng CC, Wu LW, Huang KF, Chou CY. Thrombospondin-1 acts as a fence to inhibit angiogenesis that occurs during cervical carcinogenesis. Cancer J. 2004;10:27–32. | |
Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. | |
Kim MK, Kim HS, Kim SH, et al. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem Pharmacol. 2010;80:1930–1935. | |
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. | |
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. | |
Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. | |
Peters WA 3rd, Liu PY, Barrett RJ 2nd, et al. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J Clin Oncol. 2000;18:1606–1613. | |
Marnitz S, Dowdy S, Lanowska M, Schneider A, Podratz K, Köhler C. Exenterations 60 years after first description: results of a survey among US and German Gynecologic Oncology Centers. Int J Gynecol Cancer. 2009;19:974–977. | |
Tran PT, Su Z, Hara W, Husain A, Teng N, Kapp DS. Long-term survivors using intraoperative radiotherapy for recurrent gynecologic malignancies. Int J Radiat Oncol Biol Phys. 2007;69:504–511. | |
Thigpen T, Shingleton H, Homesley H, Lagasse L, Blessing J. Cisplatinum in treatment of advanced or recurrent squamous cell carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Cancer. 1981;48:899–903. | |
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23: 1011–1027. | |
Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. | |
Kandel J, Bossy-Wetzel E, Radvanyi F, Klagsbrun M, Folkman J, Hanahan D. Neovascularization is associated with a switch to the export of bFGF in the multistep development of fibrosarcoma. Cell. 1991;66: 1095–1104. | |
Traini S, Piccolo E, Tinari N, et al. Inhibition of tumor growth and angiogenesis by SP-2, an anti-lectin, galactoside-binding soluble 3 binding protein (LGALS3BP) antibody. Mol Cancer Ther. 2014;13: 916–925. | |
Napoletano C, Bellati F, Tarquini E, et al. MAGE-A and NY-ESO-1 expression in cervical cancer: prognostic factors and effects of chemotherapy. Am J Obstet Gynecol. 2008;198:993. e1–e7. | |
Fujii T, Kuwano H. Regulation of the expression balance of angiopoietin-1 and angiopoietin-2 by Shh and FGF-2. In Vitro Cell Dev Biol Anim. 2010;46:487–491. | |
Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189:824–831. | |
Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000;60:7163–7169. | |
Giavazzi R, Sennino B, Coltrini D, et al. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162:1913–1926. | |
Daniele G, Corral J, Molife LR, de Bono JS. FGF receptor inhibitors: role in cancer therapy. Curr Oncol Rep. 2012;14:111–119. | |
Evans C, Dalgleish AG, Kumar D. Review article: immune suppression and colorectal cancer. Aliment Pharmacol Ther. 2006;24: 1163–1177. | |
Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. | |
Hopfl G, Ogunshola O, Gassmann M. HIFs and tumors – causes and consequences. Am J Physiol Regul Integr Comp Physiol. 2004;286: 608–623. | |
Brown LM, Cowen RL, Debray C, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol Pharmacol. 2006;69:411–418. | |
Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. | |
Ohm JE, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–4886. | |
Bellati F, Napoletano C, Gasparri ML, et al. Current knowledge and open issues regarding bevacizumab in gynaecological neoplasms. Crit Rev Oncol Hematol. 2012;83:35–46. | |
Papamichail M, Perez SA, Gritzapis AD, Baxevanis CA. Natural killer lymphocytes: biology, development and function. Cancer Immunol Immunother. 2004;53:176–186. | |
Dikov MM, Ohm JE, Ray N, et al. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J Immunol. 2005;174:215–222. | |
Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1: 1024–1028. | |
Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–591. | |
Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. 2010;10:575–585. | |
Karamysheva AF. Mechanisms of angiogenesis. Biochemistry. 2008;73: 751–762. | |
Nasarre P, Thomas M, Kruse K, et al. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 2009;69:1324–1333. | |
White RR, Shan S, Rusconi CP, et al. Inhibition of rat corneal angiogenesis by a nuclease resistant RNA aptamer specific for angiopoietin-2. Proc Natl Acad Sci U S A. 2003;100:5028–5033. | |
Oliner J, Min H, Leal J, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004;6:507–516. | |
Holash J, Maisonpierre PC, Compton D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. | |
Zagzag D, Hooper A, Friedlander DR, et al. In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis. Exp Neurol. 1999;159:391–400. | |
Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002;99:11205–11210. | |
Zhang L, Yang N, Park JW, et al. Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res. 2003;63:3403–3412. | |
Coxon A, Rex K, Sun J. Combined treatment of angiopoietin and VEGF pathway antagonists enhances antitumor activity in preclinical models of colon carcinoma. Abstract 1113 presented at the 2008 American Association for Cancer Research annual meeting, April 12–16, 2008, San Diego, CA, USA. | |
Hughes P, Polverino A, Oliner J, Kendall R. Angiopoietin-2 antagonists of anti-angiogenic therapy. In: Marme D, Fusenig N, editors. Tumor Angiogenesis: Basic Mechanisms and Cancer Therapy. Berlin, Germany: Springer; 2007. | |
Venneri MA, De Palma M, Ponzoni M, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276–5285. | |
Du R, Lu KV, Petritsch C, et al. HIF-1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. | |
Hashizume H, Falcón BL, Kuroda T, et al. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 2010;70:2213–2223. | |
Stafl A, Mattingly RF. Angiogenesis of cervical neoplasia. Am J Obstet Gynecol. 1975;121:845–851. | |
Vici P, Mariani L, Pizzuti L, et al. Emerging biological treatments for uterine cervical carcinoma. J Cancer. 2014;5:86–97. | |
Smith-McCune KK, Weidner N. Demonstration and characterization of the angiogenic properties of cervical dysplasia. Cancer Res. 1994;54:800–804. | |
Cooper RA, Wilks DP, Logue JP, et al. High tumor angiogenesis is associated with poorer survival in carcinoma of the cervix treated with radiotherapy. Clin Cancer Res. 1998;4:2795–2800. | |
Rubatt JM, Darcy KM, Hutson A, et al. Independent prognostic relevance of microvessel density in advanced epithelial ovarian cancer and associations between CD31, CD105, p53 status, and angiogenic marker expression: a Gynecologic Oncology Group study. Gynecol Oncol. 2009;112:469–474. | |
Cheifetz S, Bellón T, Calés C, et al. Endoglin is a component of the transforming growth factor-receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. | |
Vici P, Mariani L, Pizzuti L, et al. Immunologic treatments for precancerous lesions and uterine cervical cancer. J Exp Clin Cancer Res. 2014;33:29. | |
Zijlmans HJ, Fleuren GJ, Hazelbag S, et al. Expression of endoglin (CD105) in cervical cancer. Br J Cancer. 2009;100:1617–1626. | |
Monk BJ, Sill MW, Burger RA. Phase II trial of bevacizumab in the treatment of recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;108:S23. | |
Randall LM, Monk BJ, Darcy KM, et al. Markers of angiogenesis in high-risk, early-stage cervical cancer: A Gynecologic Oncology Group study. Gynecol Oncol. 2009;112:583–589. | |
Obermair A, Wanner C, Bilgi S, et al. Tumor angiogenesis in stage IB cervical cancer: correlation of microvessel density with survival. Am J Obstet Gynecol. 1998;178:314–319. | |
West CM, Cooper RA, Loncaster JA, Wilks DP, Bromley M. Tumor vascularity: a histological measure of angiogenesis and hypoxia. Cancer Res. 2001;61:2907–2910. | |
Kainz C, Speiser P, Wanner C, et al. Prognostic value of tumour microvessel density in cancer of the uterine cervix stage IB to IIB. Anticancer Res. 1995;15:1549–1551. | |
Rutgers JL, Mattox TF, Vargas MP. Angiogenesis in uterine cervical squamous cell carcinoma. Int J Gynecol Pathol. 1995;14:114–118. | |
Hutchison GJ, Valentine HR, Loncaster JA, et al. Hypoxia-inducible factor 1alpha expression as an intrinsic marker of hypoxia: correlation with tumor oxygen, pimonidazole measurements, and outcome in locally advanced carcinoma of the cervix. Clin Cancer Res. 2004;10:8405–8412. | |
Dobbs SP, Hewett PW, Johnson IR, Carmichael J, Murray JC. Angiogenesis is associated with vascular endothelial growth factor expression in cervical intraepithelial neoplasia. Br J Cancer. 1997;76:1410–1415. | |
Weidner N. Intratumor microvessel density as a prognostic factor in cancer. Am J Pathol. 1995;147:9–19. | |
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. | |
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. | |
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. | |
Koukourakis MI, Papazoglou D, Giatromanolaki A, Bougioukas G, Maltezos E, Sivridis E. VEGF gene sequence variation defines VEGF gene expression status and angiogenic activity in non-small cell lung cancer. Lung Cancer. 2004;46:293–298. | |
Renner W, Kotschan S, Hoffmann C, Obermayer-Pietsch B, Pilger E. A common 936 C/T mutation in the gene for vascular endothelial growth factor is associated with vascular endothelial growth factor plasma levels. J Vasc Res. 2000;37:443–448. | |
Watson CJ, Webb NJ, Bottomley MJ, Brenchley PE. Identification of polymorphisms within the vascular endothelial growth factor (VEGF) gene: correlation with variation in VEGF protein production. Cytokine. 2000;12:1232–1235. | |
Stevens A, Soden J, Brenchley PE, Ralph S, Ray DW. Haplotype analysis of the polymorphic human vascular endothelial growth factor gene promoter. Cancer Res. 2003;63:812–816. | |
Kim YH, Kim MA, Park IA, et al. VEGF polymorphisms in early cervical cancer susceptibility, angiogenesis, and survival. Gynecol Oncol. 2010;119:232–236. | |
Willmott LJ, Monk BJ. Cervical cancer therapy: current, future and anti-angiogenesis targeted treatment. Expert Rev Anticancer Ther. 2009;9:895–903. | |
Nakamura M, Bodily JM, Beglin M, Kyo S, Inoue M, Laimins LA. Hypoxia specific stabilization of HIF-1alpha by human papilloma viruses. Virology. 2009;387:442–448. | |
Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol Oncol. 2011;121:230–238. | |
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. | |
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. | |
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. | |
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. | |
Robert NJ, Diéras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252–1260. | |
Chinot OL, de La Motte Rouge T, Moore N, et al. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv Ther. 2011;28:334–340. | |
Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–2483. | |
Tomao F, Papa A, Rossi L, et al. Current status of bevacizumab in advanced ovarian cancer. Onco Targets Ther. 2013;6:889–899. | |
Grothey A, Galanis E. Targeting angiogenesis: progress with anti-VEGF treatment with large molecules. Nat Rev Clin Oncol. 2009;6: 507–518. | |
Wright JD, Viviano D, Powell MA, et al. Bevacizumab combination therapy in heavily pretreated, recurrent cervical cancer. Gynecol Oncol. 2006;103:489–493. | |
Monk BJ, Sill MW, Burger RA, Gray HJ, Buekers TE, Roman LD. Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. J Clin Oncol. 2009;27:1069–1074. | |
Kikuchi Y, Takano M, Goto T, et al. Effects of weekly bevacizumab and paclitaxel/carboplatin with or without sorafenib on heavily pretreated patients with recurrent or persistent cervical cancer. J Clin Oncol. 2011;29 Suppl:Abstr 5085. | |
Takano M, Kikuchi Y, Ikeda Y, et al. Effects of weekly bevacizumab and gemcitabine/oxaliplatin with or without dasatinib on heavily pretreated patients with recurrent or persistent cervical cancer. J Clin Oncol. 2012; 30:Suppl:Abstr e15579. | |
Schefter TE, Winter K, Kwon JS, et al. A Phase II study of bevacizumab in combination with definitive radiotherapy and cisplatin chemotherapy in untreated patients with locally advanced cervical carcinoma: preliminary results of RTOG 0417. Int J Radiat Oncol Biol Phys. 2012;83:1179–1184. | |
National Cancer Institute. Bevacizumab, radiation therapy, and cisplatin in treating patients with previously untreated locally advanced cervical cancer. ClinicalTrials.gov identifier NCT00369122. Available from: http://clinicaltrials.gov/show/NCT00369122. Accessed October 8, 2014. | |
Zighelboim I, Wright JD, Gao F, et al. Multicenter phase II trial of topotecan, cisplatin and bevacizumab for recurrent or persistent cervical cancer. Gynecol Oncol. 2013;130:64–68. | |
Tewari KS, Sill MW, Long HJ, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–743. | |
Morabito A, De Maio E, Di Maio M, Normanno N, Perrone F. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist. 2006;11:753–764. | |
Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–478. | |
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. | |
O’Farrell AM, Abrams TJ, Yuen HA, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–3605. | |
Mackay HJ, Tinker A, Winquist E, et al. A phase II study of sunitinib in patients with locally advanced or metastatic cervical carcinoma: NCIC CTG Trial IND.184. Gynecol Oncol. 2010;116:163–167. | |
Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. | |
Monk BJ, Mas Lopez L, Zarba JJ, et al. Phase II, open-label study of pazopanib or lapatinib monotherapy compared with pazopanib plus lapatinib combination therapy in patients with advanced and recurrent cervical cancer. J Clin Oncol. 2010;28:3562–3569. | |
Monk BJ, Pandite LN. Survival data from a phase II, open-label study of pazopanib or lapatinib monotherapy in patients with advanced and recurrent cervical cancer. J Clin Oncol. 2011;29:4845. | |
Diaz-Padilla I, Siu LL. Brivanib alaninate for cancer. Expert Opin Investig Drugs. 2011;20:577–586. | |
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. | |
Tomao F, Papa A, Rossi L, et al. Beyond bevacizumab: investigating new angiogenesis inhibitors in ovarian cancer. Expert Opin Investig Drugs. 2014;23:37–53. | |
Cai ZW, Zhang Y, Borzilleri RM, et al. Discovery of brivanib alaninate ((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yloxy)propan-2-yl)2 aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS-540215). J Med Chem. 2008;51:1976–1980. | |
Liang G, Chen G, Wei X, Zhao Y, Li X. Small molecule inhibition of fibroblast growth factor receptors in cancer. Cytokine Growth Factor Rev. 2013;24:467–475. | |
Bhide RS, Lombardo LJ, Hunt JT, et al. The antiangiogenic activity in xenograft models of brivanib, a dual inhibitor of vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinases. Mol Cancer Ther. 2010;9:369–378. | |
Bhide RS, Cai ZW, Zhang YZ, et al. Discovery and preclinical studies of (R)-1-(4-(4-fluoro-2-methyl-1hindol-5yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6- yloxy)propan-2-ol(BMS 540215), an in vivo active potent VEGFR-2 inhibitor. J Med Chem. 2006;49:2143–2146. | |
Gynecologic Oncology Group. Brivanib in treating patients with persistent or recurrent cervical cancer. Available from: Clinicaltrials.gov identifier NCT01267253. Accessed October 9, 2014. | |
Candelaria M, Arias-Bonfill D, Chavez-Blanco A, et al. Lack in efficacy for imatinib mesylate as second-line treatment of recurrent or metastatic cervical cancer expressing platelet-derived growth factor receptor alpha. Int J Gynecol Cancer. 2009;19:1632–1637. | |
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. | |
Shim WS, Teh M, Bapna A, et al. Angiopoietin 1 promotes tumor angiogenesis and tumor vessel plasticity of human cervical cancer in mice. Exp Cell Res. 2002;279:299–309. | |
Coxon A, Bready J, Min H, et al. Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: implications for AMG 386, an angiopoietin-1/2-neutralizing peptibody. Mol Cancer Ther. 2010;9:2641–2651. | |
Lu JF, Rasmussen E, Karlan BY, et al. Exposure response relationship of AMG386 in combination with weekly paclitaxel in recurrent ovarian cancer and its implication for dose selection. Cancer Chemother Pharmacol. 2012;69:1135–1144. | |
Monk BJ, Proveda A, Vergote I, et al. Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): a randomized, multicentre, double-bind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15:799–808. | |
Bamias A, Pignata S, Pujade-Lauraine E. Angiogenesis: a promising therapeutic target for ovarian cancer. Crit Rev Oncol Hematol. 2012;84:314–326. | |
Van Cutsem E, Tabernero J, Lakomy R, et al. Intravenous (IV) aflibercept versus placebo in combination with irinotecan/5-FU (FOLFIRI) for second-line treatment of metastatic colorectal cancer (MCRC): results of a multinational phase III trial (EFC10262-VELOUR). Presented at the 13th World Congress on Gastrointestinal Cancer, June 22–25, 2011, Barcelona, Spain. | |
Peeters M, Strickland AH, Lichinitser M, et al. A randomized, double-blind, placebo-controlled phase 2 study of trebananib (AMG 386) in combination with FOLFIRI in patients with previously treated metastatic colorectal carcinoma. Br J Cancer. 2013;108:503–511. | |
Tan SJ, Juan YH, Fu PT, et al. Chemotherapy with low-dose bevacizumab and carboplatin in the treatment of a patient with recurrent cervical cancer. Eur J Gynaecol Oncol. 2010;31(3):350–353. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.