")
Back to Journals » International Journal of General Medicine » Volume 7
An infant with poor weight gain and hypochloremic metabolic alkalosis: a case report
Authors Alhammadi A, Khalifa M, Alnaimi L
Received 20 April 2014
Accepted for publication 15 May 2014
Published 25 July 2014 Volume 2014:7 Pages 389—391
DOI https://doi.org/10.2147/IJGM.S66550
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ahmed H Alhammadi, Mohamed Khalifa, Lolwa Alnaimi
Department of Pediatrics, Division of General Pediatrics, Hamad Medical Corporation, Doha, Qatar
Abstract: Bartter syndrome is an autosomal recessive disease manifested by a defect in chloride transport in the thick loop of Henle, with different genetic origins and molecular pathophysiology. Children with Bartter syndrome generally present in early infancy with persistent polyuria and associated dehydration, electrolyte imbalance, and failure to thrive. Although early diagnosis and appropriate treatment of Bartter syndrome may improve the outcome, some children will progress to renal failure. We report a case of an 8-week-old infant who was admitted for electrolyte imbalance and failure to thrive. Laboratory studies revealed hypochloremic metabolic alkalosis with severe hypokalemia. Health care providers should consider Bartter syndrome when excessive chloride losses appear to be renal in origin and the patient has normal blood pressure and high levels of serum renin and aldosterone. Treatments, including indomethacin, spironolactone, and aggressive fluid and electrolyte replacement, may prevent renal failure in children with Bartter syndrome. Molecular genetics studies are indicated to identify the primary genetic defect.
Keywords: Bartter syndrome, failure to thrive, metabolic alkalosis
Introduction
Bartter syndrome is an autosomal recessive disease manifested by a defect in chloride transport in the thick ascending loop of Henle, known to have different genetic origins and molecular pathophysiology. However it shares the same clinical feature of failure to thrive, dehydration, and biochemical findings of renal salt wasting, metabolic alkalosis, hypokalemia, and hypercalciuria, with normal serum magnesium values.
Case presentation
An 8-week-old female infant presented to the emergency department (ED) with a 5-day history of intermittent nonbilious vomiting. The initial character of the emesis was consistent with gastroesophageal reflux, but subsequently, it became more forceful.
Prior to presentation, the infant, who was exclusively breastfed, was hungry and eager to feed between episodes of vomiting. There was no history of fever, upper respiratory tract symptoms, diarrhea, or rash, and no known contact with sick individuals. On the day of presentation, the parents reported that the infant was becoming less interested in feeding and required frequent stimulation to remain awake. Nonetheless, she was still voiding well. In general, she was less active than normal. The infant had been born to a healthy 38-year-old G3P2 mother, at 38 weeks gestational age. The pregnancy was unremarkable apart from some mild polyhydramnios noted on antenatal ultrasound. Her birth weight was 3.7 kg (50th–75th percentile). There was no other significant past medical history; the family history was also unremarkable. Furthermore, there were no developmental concerns.
On initial exam in the ED, the patient was determined to be moderately dehydrated. Following fluid resuscitation with 20 mL/kg normal saline, the patient became more alert. Her blood pressure was 80/65 mmHg, and her heart rate was regular at 140 beats per minute. At this point, the patient’s weight was 3.85 kg (fifth percentile), and she was nondysmorphic in appearance. The patient’s head and neck, respiratory, and cardiovascular exams were unremarkable. Her abdomen was soft and mildly distended, with no palpable masses and no hepatosplenomegaly. The remainder of her examination yielded no other abnormal findings.
Initial serum biochemistry revealed the following: potassium 2 mmol/L, sodium 131 mmol/L, chloride 57 mmol/L, bicarbonate 51 mmol/L, and glucose 7.1 mmol/L. Blood urea nitrogen was 1.4 mmol/L, and her creatinine was 26.5 μmol/L. Calcium and magnesium levels were normal. Complete blood count was normal, apart from a mild thrombocytosis of 470×10/L. The patient’s venous blood gas values were pH 7.59, pCO2 of 52 mmHg, pO2 50 mmHg, and HCO3 50 mmol/L, with a base excess of +15 mEq/L, consistent with metabolic alkalosis. An ultrasound of the abdomen was normal; specifically, there was no evidence of hypertrophic pyloric stenosis. Further evaluation enabled a full diagnosis.
Discussion
Our patient had a normal serum magnesium (0.74 mmol/L) concentration and a urinary Ca2+/Cr ratio of 0.98 (95th percentile for this age is 0.7), suggestive of Bartter syndrome. An elevated plasma renin level of 126.6 ng/L/s (0.13–90 ng/L/s) and an elevated aldosterone level of 2,900 pmol/L (<444 pmol/L) further supported this diagnosis. Our patient was diagnosed with Bartter syndrome type 1 (previously called the neonatal variant of Bartter syndrome). This autosomal recessive disease is manifested by a defect in chloride transport in the loop of Henle.1 The first clue to Bartter syndrome is typically maternal polyhydramnios, which can lead to preterm delivery. These children, in early infancy, generally present with persistent polyuria and associated dehydration, electrolyte imbalance, and failure to thrive. Although early diagnosis and appropriate treatment of Bartter syndrome may improve the outcome, some children will progress to renal failure.
A number of conditions should be considered in infants with persistent hypochloremic metabolic alkalosis (Table 1). Generally, this metabolic derangement may occur from either insufficient chloride intake or excessive chloride wasting. Inadequate chloride intake is a rare cause of hypochloremic metabolic alkalosis but has been described in individuals using chloride-deficient infant formulas. Inordinate amounts of chloride may be lost via gastrointestinal waste, sweat, and urine.
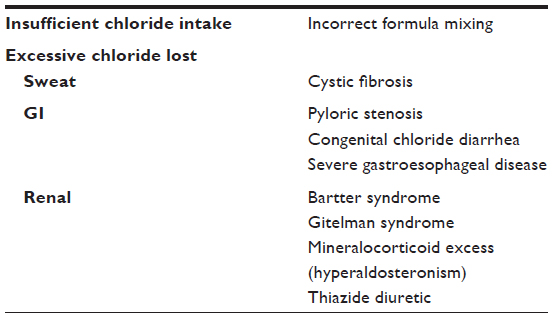 | Table 1 Differential diagnosis of hypochloremic metabolic alkalosis in infants and children |
Gastrointestinal losses of chloride may occur from excessive vomiting/suctioning of gastric secretions or from diarrhea. With an incidence of one to eight per 1,000 live births, hypertrophic pyloric stenosis should be ruled out in any infant with vomiting and hypochloremic metabolic alkalosis. The classic presentation of “projectile, nonbilious vomiting” may be a symptom in a small proportion of hypertrophic pyloric stenosis cases.2 An abdominal ultrasound, the preferred diagnostic tool for hypertrophic pyloric stenosis,3 was negative in our patient.
Excessive chloride wasting in the stool may occur secondarily to congenital chloride diarrhea, a rare autosomal recessive condition resulting in electrolyte malabsorption. Diagnosis of this condition is typically made by finding a high fecal chloride concentration (>90 mmol/L).4 Individuals with this condition require lifelong fluid and electrolyte replacement therapy.
Hypochloremic metabolic alkalosis in infants should also prompt one to consider a diagnosis of cystic fibrosis (CF). As a result of excess salt loss in sweat, hyponatremia often accompanies electrolyte imbalance in CF patients.5 In the absence of classic respiratory or gastrointestinal symptoms, CF may be suggested in early infancy, based on failure to thrive, lethargy, and dehydration due to the degree of gastrointestinal losses. Even in cases diagnosed as negative for CF, a sweat chloride test should be obtained in any infant with signs and symptoms suggestive of this disease.6
Renal causes of chloride wasting may include prolonged use of thiazide diuretics, excessive mineralocorticoid syndromes, and renal tubular losses resulting from Bartter or Gitelman syndrome. A patient’s blood pressure can help to distinguish between primary mineralocorticoid excess, which causes hypertension, and Bartter or Gitelman syndrome, in which patients remain normotensive. Our patient maintained a normal blood pressure throughout her admission.
To differentiate between Bartter and Gitelman syndromes, two genetic causes of potassium-losing nephropathies, attention should be paid to the serum magnesium and urine calcium levels. In Bartter syndrome, the serum magnesium level is usually normal, whereas the urine calcium is elevated, sometimes causing nephrocalcinosis. In Gitelman syndrome, the serum magnesium is low, and the urinary calcium level is low.
Initial management focused on correcting the fluid and electrolyte imbalance. Despite relatively good oral intake of formula, the patient showed signs of moderate dehydration. Repeat serum chemistry over the next 48 hours demonstrated persistent hypokalemia (2.5–3 mmol/L), hypochloremia (70–85 mmol/L), and low serum osmolality (260 mmol/kg H2O). Urine chemistry revealed significantly elevated chloride (61 mmol/L) and potassium (22 mmol/L) levels relative to the deficiencies of these electrolytes in the blood.
Once stabilized, our patient was maintained on oral potassium and sodium supplementation, as well as indomethacin and spironolactone, two drugs that may be helpful for this condition. Several outpatient visits at the general pediatrics and nephrology clinics have shown a well-hydrated, thriving infant, with no evidence of nephrocalcinosis on serial ultrasound.
Conclusion
In early infancy, a handful of diseases may present with hypochloremic metabolic alkalosis (Table 1).7 Although Bartter syndrome is a rare condition, health care providers should consider this when excessive chloride losses appear to be renal in origin and the patient has normal blood pressure and high levels of serum renin and aldosterone.
Treatments, including indomethacin, spironolactone, and aggressive fluid and electrolyte replacement, may prevent renal failure in children with Bartter syndrome.
Molecular genetics studies are indicated to identify the primary genetic defect.
Disclosure
The authors report no conflicts of interest in this work.
References
Proesmans W. Threading through the mizmaze of Bartter syndrome. Pediatr Nephrol. 2006;21(7):896–902. | |
Piroutek MJ, Brown L, Thorp AW. Bilious vomiting does not rule out infantile hypertrophic pyloric stenosis. Clin Pediatr (Phila). 2012;51(3):214–218. | |
Hernanz-Schulman M. Infantile hypertrophic pyloric stenosis. Radiology. 2003;227(2):319–331. | |
Hihnala S, Höglund P, Lammi L, Kokkonen J, Ormälä T, Holmberg C. Long-term clinical outcome in patients with congenital chloride diarrhea. J Pediatr Gastroenterol Nutr. 2006;42(4):369–375. | |
Ballestero Y, Hernandez MI, Rojo P, et al. Hyponatremic dehydration as a presentation of cystic fibrosis. Pediatr Emerg Care. 2006;22(11):725–727. | |
Farrell PM, Rosenstein BJ, White TB, et al; Cystic Fibrosis Foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153(2):S4–S14. | |
Chadha V, Alon US. Hereditary renal tubular disorders. Semin Nephrol. 2009;29(4):399–411. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.