")
Back to Journals » Clinical Ophthalmology » Volume 8
Advances in gene therapy technologies to treat retinitis pigmentosa
Authors Petrs-Silva H, Linden R
Received 26 September 2013
Accepted for publication 5 November 2013
Published 24 December 2013 Volume 2014:8 Pages 127—136
DOI https://doi.org/10.2147/OPTH.S38041
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Hilda Petrs-Silva, Rafael Linden
Institute of Biophysics, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
Abstract: Retinitis pigmentosa (RP) is a class of diseases that leads to progressive degeneration of the retina. Experimental approaches to gene therapy for the treatment of inherited retinal dystrophies have advanced in recent years, inclusive of the safe delivery of genes to the human retina. This review is focused on the development of gene therapy for RP using recombinant adenoassociated viral vectors, which show a positive safety record and have so far been successful in several clinical trials for congenital retinal disease. Gene therapy for RP is under development in a variety of animal models, and the results raise expectations of future clinical application. Nonetheless, the translation of such strategies to the bedside requires further understanding of the mutations and mechanisms that cause visual defects, as well as thorough examination of potential adverse effects.
Keywords: retinitis pigmentosa, photoreceptor, gene therapy, AAV
Introduction
The major causes of blindness are associated with malfunction and/or degeneration of retinal cells, which impairs the flow of visual information from the eye to the brain. Similar to other areas of the mammalian central nervous system, neurons of the retina are not replaced following degeneration. However, worldwide efforts aim to develop new therapies for preserving or improving retinal function. Such treatments are expected to slow the progression of degeneration, and if possible also to reverse its course. To help this endeavor, a number of animal models of retinal degeneration have recently led to a better understanding of mechanisms of retinal disease, and have consequently promoted the development of new therapeutic strategies.1
Diseases of the retina display remarkable genetic and clinical heterogeneity.2 Retinitis pigmentosa (RP) is the most common subtype of retinal degeneration, responsible for loss of vision in one in 4,000 people worldwide,3 one in 1,000 in the People’s Republic of China,4 and one of 930 in India.5 RP can result from defects in any of more than 60 genes inherited as autosomal dominant (30%–40% of cases), autosomal recessive (50%–60%), or X-linked (5%–15%), and it can occur either alone or together with other systemic disorders. Notwithstanding, mutations in 30%–35% of RP patients cannot be identified.6–8
Despite the heterogeneous genetic origin, RP patients display common clinical hallmarks, such as an abnormal, bone-spicule dark, punctuate pigmentation, typical of a thinned, degenerate retina; attenuated retinal vessels; dysfunction of the photoreceptors or the retinal pigment epithelium (RPE), marked by diminished electroretinogram (ERG) responses of both rods and cones, and progressive photoreceptor death. In most cases, rods are affected first, leading to night blindness, peripheral visual field loss leading to tunnel vision, and eventual total blindness. In a few cases, cones are affected first, causing loss of central vision. All RP conditions are progressive, but the speed and the pattern of deterioration of sight varies among patients. In fact, the same gene mutation can cause variable symptoms depending on the environment.6
Knowledge of underlying mechanisms of disease supports the design of optimal therapies. Such is the case with gene therapy, the aim of which is either to slow down or stop the progress of retinal degeneration in RP. Since the nature of the underlying mutation narrows down the range of treatment options, gene therapy for RP is discussed below according to the genetic classes of the disease.
Gene therapy for eye conditions
Currently, gene therapy represents the most promising therapeutic option for many inherited and acquired retinal diseases, and many preclinical and clinical assays have been done using gene therapy strategies. Recombinant adenoassociated virus (rAAV) is the most widely used vector for ocular gene delivery, because of its ability to transduce various retinal cell types in vivo efficiently,9 a result likely due to its small size relative to other viral vectors. Other advantages of AAV are the lack of pathogenicity and the ability to transduce both dividing and nondividing cells. While recombinant AAV vectors do not contain viral gene sequences, a neutralizing antibody response may be mounted against capsid proteins, which may impact the use of this vector in certain settings.10
The eye is arguably more amenable to gene therapy than other organs for several reasons: the structure and accessibility of the retina allow local, relatively noninvasive administration of the agent compared to other internal organs; treatment outcomes can be easily monitored both objectively and subjectively by noninvasive methods, such as electroretinography and optical coherence tomography, in addition to patient input; and the enclosed eye and the presence of the blood–retinal barrier prevent the unintentional systemic spread of vectors, and confer partial immune privilege status to the eye, thus limiting immune responses toward the transgene and the vector proteins.11 Notwithstanding these features, rAAV vectors are subject to intense research to improve their efficacy in gene therapy.
Ideally, therapeutic gene modulation should be restricted to specific cell types. The penetration of AAV vectors can be limited by the site of injection for intravitreal injections allow targeting to the ganglion cell layer, while subretinal injections target photoreceptors and RPE. In addition, distinct AAV-vector serotypes vary in both their targeting and transduction efficiency (Table 1).
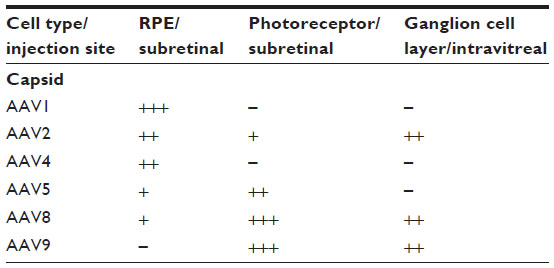 | Table 1 Efficiency of transduction of different retinal cells following subretinal or intravitreal injection of different serotypes of AAV vectors in mice |
Serotype tropism may also vary among distinct species. In canine models, various serotypes have been shown to transduce the outer retina, but recent work tends to favor especially serotypes 5 and 8 for the direct targeting of photoreceptors.12–23 In nonhuman primates, AAV2 shows good transduction in ganglion cells of the foveal area,24 rod photoreceptors and RPE25; AAV5 transduces primarily rods,26 while primate cones may be targeted by AAV5 in combination with cone-specific promoters.27 More recent work in primates showed effective transduction of photoreceptors with serotypes 1, 5, 8, and 9, with the latter showing particularly good transduction of cones.28–31 In several species, including primates, serotype 4 was shown to effectively target RPE.14
Tyrosine mutations in the capsid of AAV prevent vector ubiquitination and consequent degradation.32 New vectors have been developed in which the capsids of various AAV serotypes contain substitutions of phenylalanine for tyrosine residues. These provide for an increased efficiency of transduction, therefore reducing the amount of viral vector required for therapeutic effects, and consequently decreasing immune responses to the vector itself. Such substitutions result in increased penetration of the retina following intravitreal injections, which allows for the targeting of photoreceptors and RPE while avoiding the trauma of subretinal injections currently used to transduce these cell types.33,34
The development of AAV vectors with cell-specific promoters helps in targeting the cell of interest. rAAV vectors containing a human rpe65 promoter were used to induce RPE-specific expression in RPE65-deficient Briard dogs. This was shown to be 10% stronger then the ubiquitous cytomegalovirus promoter, and was ineffective in older animals.17 More recently, RP guanosine triphosphatase (GTPase) regulator (RPGR) promoter region was characterized, and it may be useful in future RPE targeting.35 Photoreceptors have been successfully targeted by both rhodopsin and rhodopsin-kinase promoters, with substantial activity in mice, dogs, and nonhuman primates,30,36,37 and a cone arrestin promoter has been used more recently for cone dystrophy.38,39 Promoters selective for retinal bipolar cells and ganglion cells have also been explored, mostly for optogenetic strategies of intervention through gene therapy, aimed at examining their use for recovery of visual function in patients at advanced stages of retinal degeneration.24,40
Autosomal-recessive RP
In autosomal-recessive RP, the patient has two dysfunctional copies of the mutated gene. In this case, gene-replacement therapy constitutes a straightforward approach to treat both the defective genotype and phenotype. A key element of this strategy is that the therapy is directed at the retinal cells where the mutation or lack of the relevant gene causes the primary defect. Target cells are usually photoreceptors or the RPE.
One example of gene therapy for RP in animal models that mimics the human disorder is directed at mutations in the MERTK gene. Human receptor tyrosine kinase MER (MERTK) was originally cloned as a novel tyrosine kinase,41 expressed as a transmembrane protein with two fibronectin type III domains, two immunoglobulin-like C2-type domains, and one tyrosine-kinase domain.41 In addition to its potential onco-transforming ability, deletion of the MERTK gene was identified as the underlying defect in a classic rat model of RP: the Royal College of Surgeons (RCS) rat.42 Mutations in MERTK are responsible for a rare autosomal-recessive form of RP in humans.43–45
Photoreceptors are exposed to intense levels of light that lead to the accumulation of photo-oxidized proteins and lipids, as well as free radicals, especially at the tips of the outer segments. Thus, photoreceptors have evolved to undergo constant outer-segment loss at their tips through RPE phagocytosis, together with renewal at their base via the cilium.46,47 Shed outer segments are digested in the RPE, from where important molecules are recycled to photoreceptors. Failure to regulate these functions properly can lead to the accumulation of debris in the interphotoreceptor space and retinal/RPE degeneration. MERTK located in the RPE mediates the association between these cells and the photoreceptor outer segments, and in MERTK-knockout mice, as well as in the RCS rat, in which a truncated, nonfunctional MERTK fails to localize to the cell membrane, the normal ingestion by RPE cells of the shed tips of photoreceptor outer segments is impaired. The clearance of apoptotic cells by mononuclear phagocytes is also altered in MERTK-knockout mice.48 These data are consistent with the function of MERTK in the cytoskeletal remodeling required for engulfment during phagocytosis.49
Attempts to treat MERTK defects have thus far focused on gene transfer. It was reported that transfer of normal copies of the MERTK gene by adenoviral vectors into the subretinal space of RCS rats led to both histological and functional improvement 30 days after injection. Importantly, this included correction of RPE phagocytosis defects in areas near the injection site. However, the survival of photoreceptors appeared to be only transient.50 Such transient phenotypic rescue with first-generation adenovirus vectors has been attributed to the immune response generated against viral gene products.51 Indeed, although the success of ocular gene therapy is credited in part to the relative immune-privileged status of the eye, a significant cellular immune response is known to be promoted by adenoviral proteins, which limits adenoviral-mediated transgene expression in the retina. Even though most of the adenoviral genome has been deleted in new generations of adenoviral vectors, this cellular immune response still represents a risk in the eye.52 Notwithstanding, these gene-transfer experiments with adenoviral vectors validated both that the disease phenotype is caused by mutations in MERTK and that the condition does respond to gene-replacement therapy.
In a later study, an AAV vector was used to transfer MERTK into the subretinal space of RCS rats, leading to restoration of phagocytic function, with a decrease in outer-segment debris. ERG analysis indicated transiently improved visual function and retinal morphology. The survival of photoreceptors was, however, prolonged for only 12 weeks, even in the presence of continued MERTK transgene expression.53 One possible explanation is that once the degeneration machinery is triggered, it can be delayed but not prevented.54 Since standard AAV-mediated transgene expression peaks at approximately 3–4 weeks posttreatment,55 buildup of outer-segment debris may trigger photoreceptor degeneration before peak therapeutic activity. In addition, a single subretinal injection does not cover the entire area of the retina, and degenerating photoreceptors in distant parts of the retina may have a negative impact on photoreceptor survival in the treated area, thus further limiting the effects of the treatment.
Despite the incomplete photoreceptor rescue in early studies employing rAAV vectors, the results support gene therapy as a valid therapeutic approach, as long as faster-onset vectors are used. Indeed, when a lentiviral vector was used to transfer MERTK, functional improvements lasted up to 27 weeks, and photoreceptor-cell survival was prolonged for up to 30 weeks.56
The most recent attempt to transfer MERTK via gene therapy took advantage of an AAV vector containing Y733F tyrosine-to-phenylalanine substitutions. This had been shown to provide rapid and efficient reporter-gene expression when injected subretinally into adult mouse eyes.33 The rAAV-MERTK vector led to longer and more robust functional and morphological rescue than previous studies.57
Autosomal-dominant RP
Only one mutated copy of the gene suffices to produce autosomal-dominant RP. Disease may be caused by reduction in the level of wild-type protein (haploinsufficiency), by a gain of a deleterious function (dominant negative effect), or by a combination of both. Dominant mutations may also lead to disease by causing the buildup of toxic proteins. While limited mechanistic insight has been gained from human patients, transgenic and targeted expression studies in animal models have been useful to distinguish among various types of dominant mutations. Because of the dominant nature of this class of disease, simple gene-replacement therapy is often insufficient to overcome the expression of the mutant allele, although haploinsufficiency disease may respond to gene-replacement therapy. Rather, gene therapy aimed at dominant diseases requires either suppression of the expression of the mutated allele or an increase in the expression of the wild-type allele, or both. Because there are often many disease-causing dominant mutations in a single gene, targeted gene elimination or repair for each separate mutation is problematic. An alternative approach would be to promote cell survival, to preserve affected retinal cells and slow the course of degeneration.
The most common mutations associated with autosomal dominant RP are in either the RHO or the RDS/peripherin gene, which account for approximately 25% and 10% of the cases, respectively.58 Gene therapy approaches for each are presented below.
Rhodopsin RP
The first mutation described for RP was in the rhodopsin gene – RHO. Rhodopsin is the visual pigment in rod-photoreceptor cells, which subserves vision under dim light conditions, and is involved in the essential first step of phototransduction. It consists of a protein moiety – an opsin – and a nonprotein moiety: the chromophore 11-cis-retinal. Opsin is a seven transmembrane domain-containing protein of the family of G-protein-coupled receptors, localized predominantly in the disk membranes of rod outer segments. Isomerization of 11-cis-retinal to all-trans-retinal upon absorption of a photon induces changes in opsin structure that promotes the activation of the G protein transducin, thus initiating the biochemical cascade known as phototransduction.59 Rhodopsin accounts for >70% of the total rod outer-segment protein, and more than 120 mutations located in all three domains of rhodopsin – intradiskal, transmembrane, and cytoplasmic – are associated with RP. Almost all mutations lead to the production of aberrant protein.60
The first rhodopsin mutation to be identified encoded a proline-to-histidine substitution at position 23 (P23H).61,62 P23H rhodopsin mutants are retained in the endoplasmic reticulum and are unable to associate with 11-cis-retinal.63–65 Unlike wild-type rhodopsin, mutant P23H is degraded by the ubiquitin–proteasome system,66 but large quantities of unfolded, mutant protein accumulate as ubiquitinated P23H in the cytoplasm.67 Similar to other dominant inherited neurodegenerative diseases, such as Parkinson’s and amyotrophic lateral sclerosis, the formation of intracellular protein aggregates associate with cellular degeneration.68 Mice heterozygous for RHO have normal retinal morphology and function, showing that the expression of just one functional rhodopsin allele is sufficient for vision. However, in many cases of RP, the dominant gain of function of misfolded rhodopsin induces degeneration of photoreceptors, and in such cases, mutations of one allele only lead to visual impairment. A plausible approach for the treatment of gain-of-function mutations is to enhance proteosomal degradation of misfolded rhodopsin, but to date there have been no reports of significant success in animal models. An alternative procedure might be through targeted ribonucleic acid (RNA)-based therapy to silence the mutant allele, while maintaining the expression of the wild-type allele. Gene-silencing therapies based on the selective destruction of a specific messenger RNA (mRNA) have been achieved with varying success using ribozymes, and more recently by RNA interference.69–71
Ribozymes are self-cleaving RNA enzymes of approximately 30 nucleotides, naturally found in lower eukaryotes, viruses, and some bacteria. Their secondary structure is composed of three stems: the central stem is the catalytic domain responsible for the cleavage reaction, while the two flanking domains provide the antisense arms required for mRNA binding that leads to sequence-specific cleavage.72 It was demonstrated that in vivo expression of an AAV-delivered ribozyme, designed to recognize and cleave the unique transcript produced by the P23H RHO transgene in rats, specifically reduced mRNA from the mutant allele, slowed the degeneration of photoreceptors, and led to functional preservation of the retina in the short term.73 In later studies, it was shown that the continued expression of the ribozyme markedly slowed the rate of photoreceptor degeneration and preserved retinal function, as assessed by ERG, for at least 8 months in transgenic rats.74,75 These results were the first evidence that gene-silencing approaches for disease correction can be effective for long-term therapy in autosomal-dominant retinal degeneration. However, designing and testing such therapeutic reagents for the more than 120 different rhodopsin mutations presently known is not economically or technically viable. An alternative experiment was therefore designed to attack all rhodopsin mRNAs, wild-type and mutant, at once, with a silencing agent that recognizes a common target sequence while simultaneously delivering a replacement copy of RHO, the sequence of which is resistant to the action of the silencing agent.76
More recently, small interfering RNA (siRNA) has emerged as more robust and efficient than ribozymes for silencing gene expression.77 The silencing mechanism is based on ubiquitous cellular processes, which could arguably lead to more clinical success and public acceptance. RNA interference (RNAi) inhibits gene expression by degrading mRNA in a sequence-specific manner upon introduction of double-stranded RNA (dsRNA). This long dsRNA is cut into 21- to 23-mer active intermediates, the siRNAs, which are incorporated and unwound in the RNA-induced silencing complex (RISC).78 When loaded with a single-stranded siRNA, RISC binds to the complementary sequence on the mRNA and cleaves the latter between nucleotides 10 and 11 of the siRNA,79 thus initiating its degradation and inhibiting further gene expression.
In turn, the expression of functional RHO protein coded by RHO genes resistant to siRNA has been demonstrated through transgenesis, as well as by in vivo expression of the replacement gene delivered by rAAV in the presence of the targeting RNAi molecules.80 Evidence of therapeutic benefit from AAV-delivered siRNA suppression and replacement therapies was obtained in transgenic P23H mice.76,81 These results were the first to show in vivo that the combination of suppression and replacement may treat dominantly inherited RHO-linked RP despite RHO-associated mutational heterogeneity.
RDS pattern dystrophy
Peripherin/RDS (retinal degeneration slow) is a transmembrane glycoprotein that, along with an associated protein, retinal outer-segment membrane, is localized to the rim region of outer-segment disks in rods and cones.82,83 Mice carrying a mutation in this gene (RDS/RDS) constitute one of the first and best-studied models of retinal degeneration since its phenotype was described in 1978.84 This naturally occurring null mutant fails to form photoreceptor outer segments, whereas heterozygotes have a partial phenotype of short and disorganized outer segments,85 suggesting that the protein level from one wild-type allele is not sufficient to maintain outer-segment structure and retinal function. Although these phenotypes implicate a requirement of RDS for correct outer-segment disk morphogenesis and maintenance of photoreceptor outer segments, its precise structural role is not yet completely understood.
Human RDS, cloned in 1991, encodes a putative 346-amino acid protein with 92% homology to the mouse protein.86 In the same year, RDS mutations were identified in patients with autosomal dominant RP.87 Thus far, over 90 human mutations in RDS have been identified, which result in a wide phenotypic spectrum of retinal dystrophies, related not only with RP but also with a variety of macular dystrophies, particularly pattern dystrophy. A common feature of these disorders is the loss of macular (central retinal) photoreceptors, a phenotype also seen throughout the RDS−/− mouse retina. The genotypic and phenotypic heterogeneity make the extension of studies in animal models to humans uncertain. Currently, however, gene therapy appears to be the most promising approach for treating peripherin/RDS disease. Upon subretinal injection of an AAV vector containing RDS, homozygous null mice responded with increases in rhodopsin synthesis, correction of rod outer-segment formation, and restoration of visual function in the first 14 weeks following treatment.88,89 However, the treatment did not result in long-term preservation of photoreceptors,90 demonstrating the critical importance of RDS in the integrity of the photoreceptor outer segment, and suggesting that the mutation-independent suppression and replacement strategies discussed above for RHO autosomal-dominant RP may be useful here as well.
Indeed, the concept of a double gene therapy strategy of siRNA-mediated suppression of RDS together with gene replacement through AAV vectors containing siRNA was validated with the demonstration of up to 50% reduction of RDS expression together with the simultaneous expression of a siRNA-resistant replacement transcript in the retina of mice in vivo.91
X-linked RP
X-linked RP (XLRP) is one of the most severe forms of RP, characterized by early onset and rapid progression of vision loss, accounting for 6%–20% of all RP cases. So far six loci of genetic defects have been mapped in XLRP (RP2 [MIM 312600], RP3 [MIM 312610], RP6 [MIM 312612], RP23 [MIM 300424], RP24 [MIM 300155], and RP34[MIM 300605]), but only two genes were identified: RP2 and RPGR or RP3.92,93 Studies in zebrafish provided insight into the cellular functions of both RPGR/RP3 and RP2, due to the high degree of functional conservations between human genes and their orthologues.94
Mutations in the RPGR/RP3 gene account for 70% of XLRP and disease manifest in male patients with no male-to-male transmission of the phenotype. However, families with dominant inheritance and female carriers show disease symptoms of variable degree. RPGR/RP3 product is essential for cell viability, and localizes in the connecting cilia and basal bodies of rod and cone photoreceptors, with a possible role in protein transport and microtubule organization. Approximately 60% of all XLRP cases are associated with a mutation hotspot in open reading frame in ORF15 of RPGR/RP3.95,96
A potential gene therapy for RPGR/RP3 XLRP was tested in two canine models through subretinal injection of rAAV serotype 5 coding human/RP3 under human interphotoreceptor retinoid-binding protein or G-protein-coupled receptor kinase 1 promoters. Overall, the therapy was very effective, with preservation of photoreceptor nuclei and inner/outer segments. Both rod and cone photoreceptor functions were at higher levels in treated than in control eyes, thus providing proof of principle for translation to human treatment.20
In turn, disease-causing mutations in the RP2 gene account for approximately 15% of XLRP, and are spread more uniformly along the gene. Its product is believed to have a role in the trafficking of proteins to the plasma membrane and in maintaining Golgi cohesion. A majority of the mutations in RP2 are localized in the cofactor C homologous domain, and are predicted to generate a truncated protein with disrupted localization.97,98 No gene therapy has yet been directed at this gene.
Neuroprotection in RP
Among the large number of mutations associated with RP, the common characteristic of the disease is the degeneration of rod and cone photoreceptors. Evidence both of endoplasmic reticulum stress, as well as execution of cell death through apoptosis, have been identified in photoreceptor-cell death. Accordingly, two gene therapy protocols have been designed to interfere with these events, in an attempt to preserve the visual cells. Even a delay in degeneration may be important in a combination therapy, to allow the photoreceptors time to recover function as the expression of the replacement transgene increases.
Involvement of the endoplasmic reticulum stress response was identified among pathological events of RP.99 Recently it was shown that rAAV-mediated overexpression of Bip, an endoplasmic reticulum chaperone, protected photoreceptors in the P23H RHO transgenic model of autosomal-dominant RP, and might be useful for other types of RP in which the mutation is related with the accumulation of misfolded proteins.100 On the other hand, resistance to apoptosis was achieved with rAAV-mediated delivery of the XIAP gene in cultured human RPE cells,101 and its overexpression prolonged the effects of AAV-PED6β gene therapy.102 Also, preservation of photoreceptors in various models of RP was observed following rAAV delivery of various neurotrophic factors, such as glial cell line-derived neurotrophic factor, ciliary neurotrophic factor, brain-derived neurotrophic factor, and insulin-like growth factor, among others.
Future perspectives
AAV-vectored gene therapy is now common in mouse models of various human retinal diseases. A few therapies have made their way to clinical trials, but only one has advanced enough to pave the way towards clinical use, namely the ongoing Leber congenital amaurosis 2 trials.103–109 Even in this case, recent data unraveled a dissociation between functional recovery and prevention of photoreceptor-cell death.108 Further aspects currently under investigation are related to immune responses to recombinant AAV vectors, especially upon readministration,105,110 as well as the relationship of age of onset with window of opportunity for gene therapy directed at distinct forms of Leber’s congenital amaurosis.104,111
As more studies advance, we expect further expansion of the field in the next 5–10 years. Clearly, retinal gene therapies will need to be tailored to each patient, so as to optimally address the state of the degeneration at the time of treatment. Single gene defects, if treated early, will benefit the most from direct replacement of therapeutic genes or correction/ablation of the offending gene. For many such disorders, however, patient numbers are so small that more general neuroprotective gene therapies are likely to be developed first, because they hold promise for preventing or delaying degeneration in a wide variety of retinal disorders.
For late-stage retinal dystrophies, in which photoreceptor-mediated vision is essentially absent, prosthetic retinas, cell-replacement therapies or gene-based modifications of inner retinal cells to become light receptors may be the most likely alternatives.112 Stem and progenitor cells can be isolated from a number of sources, including embryonic tissue, adult brain, and even retina, prompting many researchers to investigate the potential for using these cells to generate retinal cells for transplantation.113,114 However, there are several obstacles to be overcome before these techniques can be applied, such as the poor yield of differentiation of exogenous stem cells and the complexity of the required synaptic connections between transplanted and endogenous retinal cells. Exploitation of cell replacement will require a deeper understanding of developmental biology and the identification of key regulators of the various cellular differentiation pathways.
In conclusion, the advances in the understanding of the genetics and pathophysiology of retinal disease have now established the fundamentals of new gene-based therapies for several disorders that have not responded to conventional treatments, and recombinant AAV vectors are at the center of this progress towards the cure of retinal degenerations.
Acknowledgments
The authors’ research was funded by CNPq and FAPERJ, and there are no conflicts of interest regarding this publication.
Disclosure
The authors report no other conflicts of interest in this work.
References
Fletcher EL, Jobling AI, Vessey KA, Luu C, Guymer RH, Baird PN. Animal models of retinal disease. Prog Mol Biol Transl Sci. 2011;100: 211–286. | |
Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190(6):953–963. | |
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. | |
Xu L, Hu L, Ma K, Li J, Jonas JB. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: the Beijing Eye Study. Eur J Ophthalmol. 2006;16(6):865–866. | |
Sen P, Bhargava A, George R, et al. Prevalence of retinitis pigmentosa in South Indian population aged above 40 years. Ophthalmic Epidemiol. 2008;15(4):279–281. | |
Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29(5):335–375. | |
Sahel J, Bonnel S, Mrejen S, Paques M. Retinitis pigmentosa and other dystrophies. Dev Ophthalmol. 2010;47:160–167. | |
Anasagasti A, Irigoyen C, Barandika O, Lopez de Munain A, Ruiz-Ederra J. Current mutation discovery approaches in retinitis pigmentosa. Vision Res. 2012;75:117–129. | |
Vandenberghe LH, Auricchio A. Novel adeno-associated viral vectors for retinal gene therapy. Gene Ther. 2012;19(2):162–168. | |
Alexander JJ, Hauswirth WW. Adeno-associated viral vectors and the retina. Adv Exp Med Biol. 2008;613:121–128. | |
Streilein JW. Ocular immune privilege: the eye takes a dim but practical view of immunity and inflammation. J Leukoc Biol. 2003;74(2):179–185. | |
Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28(1):92–95. | |
Narfström K, Katz ML, Ford M, Redmond TM, Rakoczy E, Bragadóttir R. In vivo gene therapy in young and adult RPE65-/- dogs produces long-term visual improvement. J Hered. 2003;94(1):31–37. | |
Weber M, Rabinowitz J, Provost N, et al. Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther. 2003;7(6):774–781. | |
Bainbridge JW, Mistry A, Schlichtenbrede FC, et al. Stable rAAV-mediated transduction of rod and cone photoreceptors in the canine retina. Gene Ther. 2003;10(16):1336–1344. | |
Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12(6):1072–1082. | |
Le Meur G, Stieger K, Smith AJ, et al. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007;14(4):292–303. | |
Komaromy AM, Alexander JJ, Cooper AE, et al. Targeting gene expression to cones with human cone opsin promoters in recombinant AAV. Gene Ther. 2008;15(14):1049–1055. | |
Beltran WA, Boye SL, Boye SE, et al. rAAV2/5 gene-targeting to rods:dose-dependent efficiency and complications associated with different promoters. Gene Ther. 2010;17(9):1162–1174. | |
Beltran WA, Cideciyan AV, Lewin AS, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109(6):2132–2137. | |
Komaromy AM, Rowlan JS, Corr AT, et al. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol Ther. 2013;21(6):1131–1141. | |
Annear MJ, Mowat FM, Bartoe JT, et al. Successful gene therapy in older Rpe65-deficient dogs following subretinal injection of an adeno-associated vector expressing RPE65. Hum Gene Ther. 2013;24(10):883–893. | |
Lhériteau E, Petit L, Weber M, et al. Successful gene therapy in the RPGRIP1-deficient dog: a large model of cone-rod dystrophy. Mol Ther. Epub October 4, 2013. | |
Yin L, Greenberg K, Hunter JJ, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci. 2011;52(5):2775–2783. | |
Bennett J, Maguire AM, Cideciyan AV, et al. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc Natl Acad Sci U S A. 1999;96(17):9920–9925. | |
Lotery AJ, Yang GS, Mullins RF, et al. Adeno-associated virus type 5: transduction efficiency and cell-type specificity in the primate retina. Hum Gene Ther. 2003;14(17):1663–1671. | |
Mancuso K, Hendrickson AE, Connor TB Jr, et al. Recombinant adeno-associated virus targets passenger gene expression to cones in primate retina. J Opt Soc Am A Opt Image Sci Vis. 2007;24(5):1411–1416. | |
Vandenberghe LH, Bell P, Maguire AM, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3(88):88ra54. | |
Baba Y, Satoh S, Otsu M, Sasaki E, Okada T, Watanabe S. In vitro cell subtype-specific transduction of adeno-associated virus in mouse and marmoset retinal explant culture. Biochimie. 2012;94(12):2716–2722. | |
Boye SE, Alexander JJ, Boye SL, et al. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum Gene Ther. 2012;23(10):1101–1115. | |
Vandenberghe LH, Bell P, Maguire AM, et al. AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS One. 2013;8(1):e53463. | |
Yan Z, Zak R, Luxton GW, Ritchie TC, Bantel-Schaal U, Engelhardt JF. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol. 2002;76(5):2043–2053. | |
Petrs-Silva H, Dinculescu A, Li Q, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17(3):463–471. | |
Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011;19(2):293–301. | |
Shu X, Simpson JR, Hart AW, et al. Functional characterization of the human RPGR proximal promoter. Invest Ophthalmol Vis Sci. 2012;53(7):3951–3958. | |
Khani SC, Pawlyk BS, Bulgakov OV, et al. AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest Ophthalmol Vis Sci. 2007;48(9):3954–3961. | |
Pang J, Boye SE, Lei B, et al. Self-complementary AAV-mediated gene therapy restores cone function and prevents cone degeneration in two models of Rpe65 deficiency. Gene Ther. 2010;17(7):815–826. | |
Pickrell SW, Zhu X, Wang X, Craft CM. Deciphering the contribution of known cis-elements in the mouse cone arrestin gene to its cone-specific expression. Invest Ophthalmol Vis Sci. 2004;45(11):3877–3884. | |
Carvalho LS, Xu J, Pearson RA, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20(16):3161–3175. | |
Doroudchi MM, Greenberg KP, Liu J, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19(7):1220–1229. | |
Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994;5(6):647–657. | |
D’Cruz PM, Yasumura D, Weir J, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9(4):645–651. | |
Gal A, Li Y, Thompson DA, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26(3):270–271. | |
Tada A, Wada Y, Sato H, et al. Screening of the MERTK gene for mutations in Japanese patients with autosomal recessive retinitis pigmentosa. Mol Vis. 2006;12:441–444. | |
Brea-Fernández AJ, Pomares E, Brión MJ, et al. Novel splice donor site mutation in MERTK gene associated with retinitis pigmentosa. Br J Ophthalmol. 2008;92(10):1419–1423. | |
Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–195. | |
Nguyen-Legros J, Hicks D. Renewal of photoreceptor outer segments and their phagocytosis by the retinal pigment epithelium. Int Rev Cytol. 2000;196:245–313. | |
Scott RS, McMahon EJ, Pop SM, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–211. | |
Jennings JH, Linderman DJ, Hu B, Sonstein J, Curtis JL. Monocytes recruited to the lungs of mice during immune inflammation ingest apoptotic cells poorly. Am J Respir Cell Mol Biol. 2005;32(2):108–117. | |
Vollrath D, Feng W, Duncan JL, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci U S A. 2001;98(22):12584–12589. | |
Reichel MB, Ali RR, Thrasher AJ, Hunt DM, Bhattacharya SS, Baker D. Immune responses limit adenovirally mediated gene expression in the adult mouse eye. Gene Ther. 1998;5(8):1038–1046. | |
Bennett J. Immune response following intraocular delivery of recombinant viral vectors. Gene Ther. 2003;10(11):977–982. | |
Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR. AAV-mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8(2):188–195. | |
Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, et al. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol. 2008;38(3):253–269. | |
Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10(26):3075–3081. | |
Tschernutter M, Schlichtenbrede FC, Howe S, et al. Long-term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther. 2005;12(8):694–701. | |
Dinculescu A, Estreicher J, Zenteno JC, et al. Gene therapy for retinitis pigmentosa caused by MFRP mutations: human phenotype and preliminary proof of concept. Hum Gene Ther. 2012;23(4):367–376. | |
Fingert JH, Oh K, Chung M, et al. Association of a novel mutation in the retinol dehydrogenase 12 (RDH12) gene with autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 2008;126(9):1301–1307. | |
Ridge KD, Palczewski K. Visual rhodopsin sees the light: structure and mechanism of G protein signaling. J Biol Chem. 2007;282(13):9297–9301. | |
Wert KJ, Davis RJ, Sancho-Pelluz J, Nishina PM, Tsang SH. Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2013;22(3):558–567. | |
Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–366. | |
Humphries P, Farrar GJ, Kenna P, McWilliam P. Retinitis pigmentosa: genetic mapping in X-linked and autosomal forms of the disease. Clin Genet. 1990;38(1):1–13. | |
Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88(15):6481–6485. | |
Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33(20):6121–6128. | |
Cideciyan AV, Hood DC, Huang Y, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998;95(12):7103–7108. | |
Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115(Pt 14):2907–2918. | |
Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277(37):34150–34160. | |
Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am J Neurodegener Dis. 2013;2(1):1–14. | |
Lewin AS, Hauswirth WW. Ribozyme gene therapy: applications for molecular medicine. Trends Mol Med. 2001;7(5):221–228. | |
Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res. 2007;84(1):44–52. | |
Mao H, Gorbatyuk MS, Rossmiller B, Hauswirth WW, Lewin AS. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum Gene Ther. 2012;23(4):356–366. | |
Gwiazda S, Salomon K, Appel B, Müller S. RNA self-ligation: from oligonucleotides to full length ribozymes. Biochimie. 2012;94(7):1457–1463. | |
Lewin AS, Drenser KA, Hauswirth WW, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4(8):967–971. | |
LaVail MM, Yasumura D, Matthes MT, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A. 2000;97(21):11488–11493. | |
Hauswirth WW, Lewin AS. Ribozyme uses in retinal gene therapy. Prog Retin Eye Res. 2000;19(6):689–710. | |
O’Reilly M, Palfi A, Chadderton N, et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. 2007;81(1):127–135. | |
Golden DE, Gerbasi VR, Sontheimer EJ. An inside job for siRNAs. Mol Cell. 2008;31(3):309–312. | |
Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404(6775):293–296. | |
Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15(2):188–200. | |
Kiang AS, Palfi A, Ader M, et al. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol Ther. 2005;12(3):555–561. | |
Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Suppression of mouse rhodopsin expression in vivo by AAV mediated siRNA delivery. Vision Res. 2007;47(9):1202–1208. | |
Molday RS, Hicks D, Molday L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Invest Ophthalmol Vis Sci. 1987;28(1):50–61. | |
Farjo R, Naash MI. The role of Rds in outer segment morphogenesis and human retinal disease. Ophthalmic Genet. 2006;27(4):117–122. | |
van Nie R, Iványi D, Démant P. A new H-2-linked mutation, rds, causing retinal degeneration in the mouse. Tissue Antigens. 1978;12(2):106–108. | |
Hawkins RK, Jansen HG, Sanyal S. Development and degeneration of retina in rds mutant mice: photoreceptor abnormalities in the heterozygotes. Exp Eye Res. 1985;41(6):701–720. | |
Travis GH, Christerson L, Danielson PE, et al. The human retinal degeneration slow (RDS) gene: chromosome assignment and structure of the mRNA. Genomics. 1991;10(3):733–739. | |
Boon CJ, den Hollander AI, Hoyng CB, Cremers FP, Klevering BJ, Keunen JE. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;27(2):213–235. | |
Ali RR, Sarra GM, Stephens C, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25(3):306–310. | |
Schlichtenbrede FC, da Cruz L, Stephens C, et al. Long-term evaluation of retinal function in Prph2Rd2/Rd2 mice following AAV-mediated gene replacement therapy. J Gene Med. 2003;5(9):757–764. | |
Sarra GM, Stephens C, de Alwis M, et al. Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina. Hum Mol Genet. 2001;10(21):2353–2361. | |
Petrs-Silva H, Yasumura D, Matthes MT, LaVail MM, Lewin AS, Hauswirth WW. Suppression of rds expression by siRNA and gene replacement strategies for gene therapy using rAAV vector. Adv Exp Med Biol. 2012;723:215–223. | |
Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70(6):1545–1554. | |
Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73(5):1131–1146. | |
Raghupathy RK, McCulloch DL, Akhtar S, Al-Mubrad TM, Shu X. Zebrafish model for the genetic basis of X-linked retinitis pigmentosa. Zebrafish. 2013;10(1):62–69. | |
Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25(4):462–466. | |
Murga-Zamalloa CA, Swaroop A, Khanna H. RPGR-containing protein complexes in syndromic and non-syndromic retinal degeneration due to ciliary dysfunction. J Genet. 2009;88(4):399–407. | |
Mears AJ, Gieser L, Yan D, et al. Protein-truncation mutations in the RP2 gene in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1999;64(3):897–900. | |
Schwahn U, Paland N, Techritz S, Lenzner S, Berger W. Mutations in the X-linked RP2 gene cause intracellular misrouting and loss of the protein. Hum Mol Genet. 2001;10(11):1177–1183. | |
Sanges D, Comitato A, Tammaro R, Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci U S A. 2006;103(46):17366–17371. | |
Gorbatyuk MS, Knox T, LaVail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107(13):5961–5966. | |
Shan H, Ji D, Barnard AR, et al. AAV-mediated gene transfer of human X-linked inhibitor of apoptosis protects against oxidative cell death in human RPE cells. Invest Ophthalmol Vis Sci. 2011;52(13):9591–9597. | |
Yao J, Jia L, Khan N, et al. Caspase inhibition with XIAP as an adjunct to AAV vector gene-replacement therapy: improving efficacy and prolonging the treatment window. PLoS One. 2012;7(5):e37197. | |
Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20(9):999–1004. | |
Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–1605. | |
Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4(120):120ra115. | |
Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest. 2011;121(6):2160–2168. | |
Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. | |
Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110(6):E517–E525. | |
Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130(1):9–24. | |
Willett K, Bennett J. Immunology of AAV-mediated gene transfer in the eye. Front Immunol. 2013;4:261. | |
Tan MH, Mackay DS, Cowing J, et al. Leber congenital amaurosis associated with AIPL1: challenges in ascribing disease causation, clinical findings, and implications for gene therapy. PLoS One. 2012;7(3):e32330. | |
Jones BW, Kondo M, Terasaki H, Lin Y, McCall M, Marc RE. Retinal remodeling. Jpn J Ophthalmol. 2012;56(4):289–306. | |
Ramsden CM, Powner MB, Carr AJ, Smart MJ, da Cruz L, Coffey PJ. Stem cells in retinal regeneration: past, present and future. Development. 2013;140(12):2576–2585. | |
Ong JM, da Cruz L. A review and update on the current status of stem cell therapy and the retina. Br Med Bull. 2012;102:133–146. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.