")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Acute kidney injury: what part do toll-like receptors play?
Authors Vallés P, Gil Lorenzo A, Bocanegra V, Vallés R
Received 19 February 2014
Accepted for publication 28 March 2014
Published 19 June 2014 Volume 2014:7 Pages 241—251
DOI https://doi.org/10.2147/IJNRD.S37891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Patricia G Vallés,1,2 Andrea Gil Lorenzo,2 Victoria Bocanegra,2 Roberto Vallés3
1Área de Fisiopatología, Departamento de Patología, Facultad de Ciencias Médicas, Universidad Nacional de Cuyo, Mendoza, Argentina; 2National Council of Scientific and Technical Research of Argentina, Buenos Aires, Argentina; 3Instituto de Inmunología Facultad de Ciencias Médicas, Universidad Nacional de Cuyo, Mendoza, Argentina
Abstract: The innate immune system plays an important role as a first response to tissue injury. This first response is carried out via germline-encoded receptors. Toll-like receptors (TLRs) are the first identified and best studied family of pattern recognition receptors. TLRs are expressed on a variety of cell types, including epithelial cells, endothelia, dendritic cells, monocytes/macrophages, and B- and T-cells. TLRs initiate innate immune responses and concurrently shape the subsequent adaptive immune response. They are sensors of both pathogens, through the exogenous pathogen-associated molecular patterns (PAMPs), and tissue injury, through the endogenous danger-associated molecular patterns (DAMPs). TLR signaling is critical in defending against invading microorganisms; however, sustained receptor activation is also implicated in the pathogenesis of inflammatory diseases. Ischemic kidney injury involves early TLR-driven immunopathology, and the resolution of inflammation is needed for rapid regeneration of injured tubule cells. Notably, the activation of TLRs also has been implicated in epithelial repair. This review focuses on the role of TLRs and their endogenous ligands within the inflammatory response of acute kidney injury.
Keywords: toll-like receptors, pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), ischemic kidney injury
Introduction
All living organisms are constantly exposed to microorganisms that are present in the environment and need to cope with invasion of these organisms into the body. The vertebrate immune response can be divided into innate and acquired immunity.
Phylogenetically conserved, the innate immune system is present in almost all multicellular organisms. This first line of defense is performed via germline-encoded receptors expressed by macrophages, lymphocytes, dendritic cells, monocytes, neutrophils, and natural killer cells, among others.1 Acquired immune responses, by contrast, are slower processes mediated by T- and B-cells, both of which express highly diverse antigen receptors that are generated through the deoxyribonucleic acid (DNA) rearrangement and are, thereby, able to respond to a wide range of potential antigens. This highly sophisticated system of antigen detection is found only in vertebrates and has been the subject of considerable research. Far less attention has been directed toward innate immunity, as it has been regarded as a relatively nonspecific system with its main roles being to destroy pathogens and to present antigen to the cells involved in acquired immunity.
Recent studies, however, have shown that the innate immune system has a greater degree of specificity than was previously thought and that it is highly developed in its ability to discriminate between self- and foreign pathogens.1 This discrimination relies, to a great extent, on a family of evolutionarily conserved receptors, known as the toll-like receptors (TLRs), which have a crucial role in early host defense against invading pathogens.1,2
Furthermore, accumulating evidence indicates that activation of the innate immune system is a prerequisite for the induction of acquired immunity, particularly for the induction of a T helper 1 (TH1)-cell response.3,4
This review summarizes the present data regarding the role of TLRs and their endogenous ligand activation in the course and development of acute kidney injury (AKI).
TLRs
The TLRs were discovered more than a decade ago as sentinel receptors for the mammalian innate immune system. The susceptibility to fungal infection in toll-deficient Drosophila, demonstrated by Lemaitre et al,5 led to the discovery of TLRs in humans in rodents. TLR are mammalian homologues of the Toll receptor, first described in Drosophila.6,7 The TLRs are among a growing number of receptors that detect danger signals in extracellular and intracellular compartments.
Several TLRs were identified and observed recognizing a wide range of pathogen-associated molecular patterns (PAMPs), including peptidoglycan, lipopolysaccharide (LPS), CpG DNA, and double-stranded or single-stranded ribonucleic acid (RNA) derived from microorganisms.8 In response, it triggers an inflammatory cascade that includes activation and maturation of dendritic cells. Dendritic cells are the most potent antigen-presenting cells of innate immunity.9 The TLR-activated dendritic cells induce naïve T lymphocytes to mature into antigen-specific effector T-cells, particularly of the TH1 lineage.2 Thus, TLRs link innate and adaptive immune responses, both of which are critical to host defense against pathogens. TLRs are also implicated in the pathogenesis of several inflammatory diseases, including kidney diseases.8
Endogenous molecules that are generated during tissue injury and labeled as damage-associated molecular pattern molecules (DAMPs) can also activate pattern recognition receptors similarly to PAMPs, thereby offering a novel understanding of sterile types of inflammation.10
Intracellular sources can originate DAMPs or they can be generated from extracellular matrix degradation. DAMPs might be generated and released during cell stress, apoptosis, or necrosis due to traumatic, ischemic, toxic, or inflammatory tissue injuries. It is likely that DAMPs function as danger signals and that DAMP-mediated immune activation developed during evolution to aid danger control and tissue repair. If such a process escapes the normal control and/or suppression of an adaptive immune response to endogenous molecules, the recognition of PAMP and DAMP by receptors of the innate immune system could contribute to an autoimmune response.11 In particular, increasing evidence suggests that endogenous ligands activate TLRs, resulting in the antigen-independent inflammation that accompanies ischemic AKI, solid organ transplant rejection, and immune-mediated glomerulonephritis.8
TLRs: structure and basic role
Up to now, eleven human and 13 mouse TLRs have been identified. All are structurally similar, highlighting – in terms of evolution – that this is an ancient system for immune protection. TLRs are transmembrane receptors, type I integral membrane glycoproteins. They contain a cytoplasmic domain named the toll/interleukin (IL)-1R (TIR) domain because of sequence homology with the IL-1 receptor (IL-1R).8 By contrast, the extracellular region of the TLRs and IL-1Rs differs markedly. The extracellular region of TLRs contains leucine-rich repeat (LRR) motifs; whereas, the extracellular region of IL-1Rs contains three immunoglobulin-like domains.12
TLRs are divided into two categories, according to their localization: 1) those found in the cell surface membrane (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10, TLR11, TLR12, and TLR13); and 2) those found primarily on endosome membranes (TLR3, TLR7, TLR8, and TLR9), these last three TLRs are involved in the recognition of nucleic-acid-like structures. The most recently discovered member of the TLR family, TLR-15, has been associated with recognition of Salmonella components.8
Compartmentalization between different types of TLRs varies. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are anchored on the cell surface and are capable of recognizing PAMPs and DAMPs derived from invading pathogens or the host, while TLR3, TLR7, TLR8, and TLR9 are localized inside the cell, primarily sensing viral, bacterial, or host-derived nucleic acids in endosomes.13 The recognition of a plethora of PAMPs and DAMPs by the ancient family of TLRs lies in their diversity (13 in mammals) and their ability to form homodimers or heterodimers induced by the simultaneous binding of ligands to residues within LRRs.14,15 Upon stimulation, TLRs recruit adaptor molecules.
TLR expression
TLRs are found on a variety of cell types, including epithelial cells, endothelia, dendritic cells, monocytes/macrophages, and B- and T-cells.16 Each cell type may express only a limited number of TLRs. For example, plasmacytoid dendritic cells are a specialized cell population that produces large amounts of type I interferon (IFN), in response to the viruses’ expression of TLR7 and TLR9, but not other TLRs.17
In the kidney, the tubular epithelial cells and mesangial cells express TLR1 through TLR4 and TLR6.18 The TLR2 protein expression has been demonstrated in many cell types in the kidney, including renal tubules of the outer stripe of the medulla, glomeruli, and in the renal vasculature. There are similarities in the pattern of protein expression in humans and mice. Data on the TLR expression in podocytes and other glomerular components are sparse, although a constitutive TLR2 staining in glomerular capillary endothelial cells has been shown.19 Most TLRs reside on the cell surface, except TLR3 and TLR7 through TLR9, which are found intracellularly to encounter ligands in endosomes or lysosomes. The wide distribution of TLRs in different tissues, cells, and subcellular compartments allow them to be well-positioned as sentinels against invading pathogens.8
TLRs signaling pathways
The cytosolic domains of TLR recruit adaptor proteins and initiate signaling pathways in response to ligand binding (Figure 1). Five adaptor proteins have been discovered: 1) myeloid differentiation factor 88 (MyD88); 2) MyD88 adaptor-like (MAL, also known as TIRAP); 3) TIR domain-containing adaptor protein-inducing IFN-β (TRIF, also known as TICAM1); 4) TRIF-related adaptor molecule (TRAM, also known as TICAM2); and 5) sterile α and armadillo motif-containing protein.
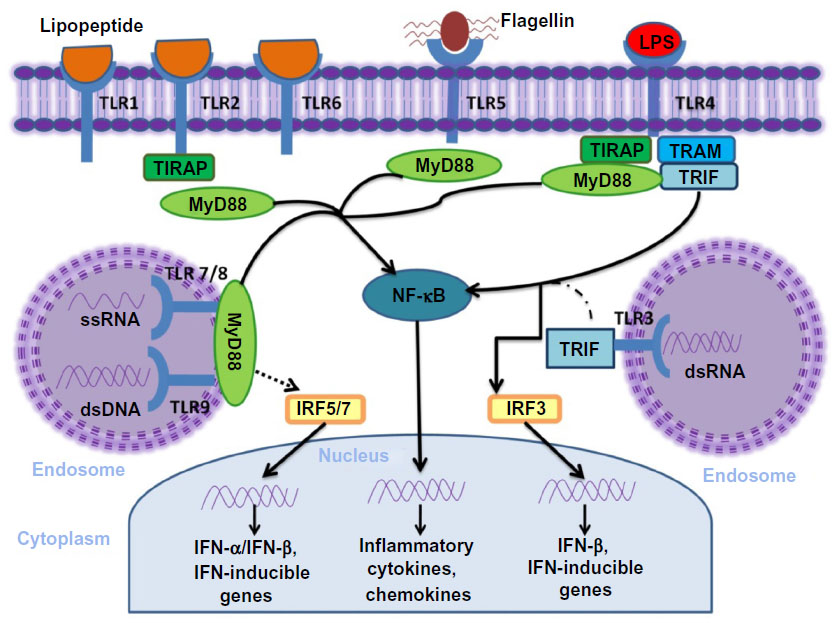 | Figure 1 TLR and their major signal adaptors. |
According to the recruited adaptor molecules, TLR1, TLR2, TLR7, and TLR9 signaling is achieved through the MyD88 pathway, TLR3 signals through the TRIF pathway; whereas, TLR2 and TLR4 recruit MyD88 through MAL/TIRAP. TLR4 also engages TRIF through TRAM.8,20 Once stimulated, the MyD88-dependent pathway leads to the activation of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) mitogen-activated protein kinase and IFN regulatory factor pathways of inflammation, cell growth, and differentiation. The TRIF-dependent pathway also activates NF-κB with delayed kinetics and additionally induces IFN regulatory protein 3, a transcription factor necessary for production of type I IFN. TLR1, TLR2, and TLR6 contain a phosphatidylinositol 3-kinase (PI3K) binding motif 1 and activate NF-κB through PI3K independent of MyD88. Both MyD88 and TRIF pathways can also lead to cell apoptosis by activating Fas-associated protein with death domain and, finally, caspase.8,21,22
The specificity of the TLRs function is determined by the selective use of these intracellular adapter molecules. Combinations of TLR ligand, cell type, receptor, and adaptor protein lead to different signaling pathways that result in a diverse range of cellular responses – all of which support a strong innate immune response.
TLR ligand signals
Response to foreign and endogenous danger signals
The immune system activation in response to foreign pathogens is recapitulated in an immune response to endogenous molecules released from necrotic and perhaps apoptotic cells after tissue injury or trauma related to hypoxia, ischemia, mechanical stress, or pathogen-induced inflammation. A variety of activators, including DNA, RNA, lipids, and peptide products from bacteria, viruses, fungi, and synthetically derived compounds are involved in the TLR response.23 LPS, the best characterized ligand, is the cell wall component of gram-negative bacteria that plays a prominent role in the pathogenesis of sepsis. LPS activates TLR4, and this interaction contributes to the inflammation that characterizes LPS-induced AKI.8,24
The TLR agonist’s identity has grown after the inclusion of the endogenous ligands. Dying cells release endogenous molecules (DAMPs) and activate cellular receptors leading to downstream inflammation (Figure 2). They are recognized as danger signals when released into the extracellular space exposing hydrophobic portions of the molecules that are normally hidden in healthy living cells. The high mobility box group 1 (HMGB1) nuclear protein, hyaluronan, and heat-shock proteins are included as endogenous ligands;25,26 these molecules activate TLRs.
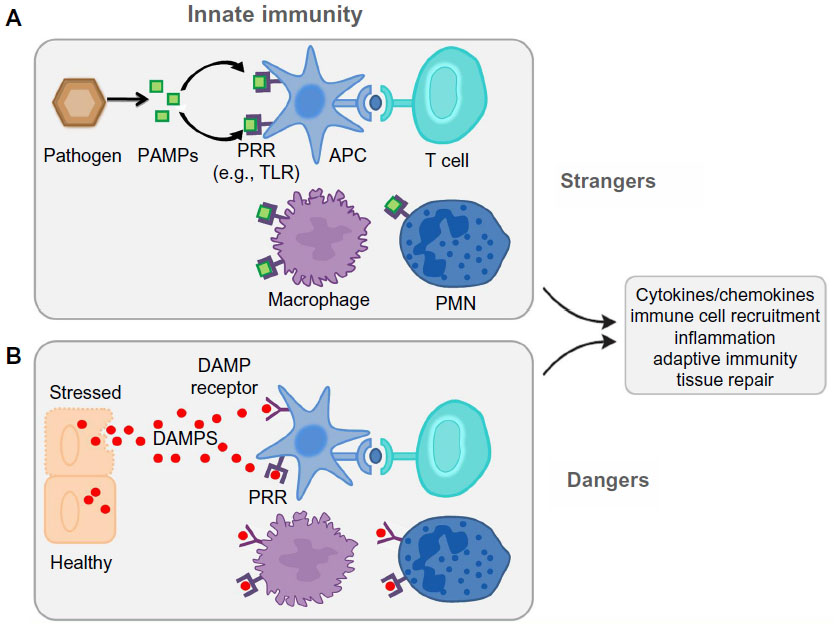 | Figure 2 Danger and stranger models. |
The traditional concept of immune surveillance as self/nonself discrimination (the stranger hypothesis) assumes that the foreignness of a particular entity is what triggers both the innate and adaptive response. In contrast, the danger model holds that the immune system is governed from within, responding to endogenous signals that originate from stressed or injured cells.27,28 The danger model assumes – from an evolutionary point of view – that what really matters is whether that entity causes damage or not. If it does not, and the cells in its environment are healthy or if they die normal quiet deaths and are scavenged, no immune response ensues. Only when a cell dies messily, or becomes stressed or damaged, is an immune response initiated. It does not matter if the damage is done by a pathogen or by an innate defect in the cell’s programming.
This basic difference leads to a different view of: 1) the origin of the signals; and 2) the nature of the cells that control immunity. The result is a quite different set of explanations for many aspects of immunity, as well as a different perspective on the evolutionary origins of the immune system.
Until now, there is more that needs to be learned about the role of danger signals in tissue injury. However, the paradigm shift in the traditional concept of immune surveillance as self/nonself discrimination (the stranger hypothesis) to the danger model, as proposed by Matzinger,29 more readily explains a role for the innate immune system in AKI organ transplantation and autoimmune disease.
Inflammatory response to AKI
A mechanistic explanation of the development of systemic inflammatory responses across a broad spectrum of nonbacterial diseases has emerged with the discovery of components of the innate immunity against pathogenic bacteria–pathogen associated molecular patterns and pathogen-recognition receptors, and one of their major components, TLRs. This bacterial defense system can be hijacked by a host of molecules, alarmins, that are usually released during inflammation. This last process has been conceptualized by Matzinger29 as the danger model.
Inflammation at the site of tissue injury is a hallmark of almost all forms of renal injury, and it is an important factor in the development of many kidney diseases. It has become clear that renal epithelium plays a crucial role in the attraction of leukocytes upon injury, at least partially in a TLR-dependent manner.30 TLR4, a member of the TLR family, is especially promiscuous with respect to potential ligands; it has been implicated in the pathogenesis of AKI.31
TLR signaling in ischemic kidney injury
AKI results most commonly from ischemic/reperfusion (I/R) injury; and, although different mechanisms are involved, a common pathway leading to proximal tubule injury is the activation of innate and adaptive immunity leading to inflammation. Renal I/R injury is a major cause of morbidity in both allograft and native kidneys, and it is most commonly associated with either transplant or trauma, leading to poor clinical outcome.32
A robust inflammatory response – triggered by hypoxia and by the process of reperfusion – determined the outcome of the ischemic organ. After necrotic death, intracellular molecules may find their way into the extracellular space and there recruit and regulate the inflammatory infiltrate. Characterized by the presence of inflammatory cytokines and chemokines, this inflammation process also includes increased expression of adhesion molecules and recruitment of leukocytes into the postischemic kidney. The major factors in regulating inflammation are necrotic and apoptotic cells. There is also strong evidence that TLR2 and TLR4 participate in this process.
Kim et al33 were the first to show the gene and protein expression upregulation of TLR2 and TLR4 in rat kidney tissues after reperfusion in ischemic injury. TLR2 and TLR4 are constitutively expressed in both proximal and distal tubules, the thin limb of the Henle loop, and in the collecting ducts. Kim et al33 also showed that, in fact, the expression of these receptors were upregulated in these areas post I/R. Wolfs et al34 found that TLR2 and TLR4 are constitutively expressed in primarily proximal and distal tubule epithelial cells and in the epithelium of Bowman’s capsule in healthy, wild-type murine kidneys. Both TLR expressions rapidly increased in ischemic kidneys, up to four- to five-fold over basal levels, especially in distal tubular epithelia, at day 5 after ischemia.34
The role of TLR2 in injury using TLR2−/− and TLR2+/+ mice, TLR2 antisense oligonucleotides, and chimeric mice deficient in leukocyte or renal TLR2 were evaluated by Leemans et al.31 After I/R injury in vivo, the TLR2 played a proinflammatory and detrimental role, supported by the results of local cytokines, chemokines, and leukocytes reduction and the decreased renal injury and dysfunction in TLR2−/− mice compared with controls. An analysis of chimeric mice suggested that TLR2 expressed on renal parenchyma plays a crucial role in the induction of inflammation and injury.
The constitutive TLR2 and TLR4 higher messenger RNA (mRNA) expressed by renal tubular epithelial cells (TECs) is enhanced upon renal I/R injury, being the activity of these receptors strongly upregulated by the endogenous ligands.35
Together, these data suggest a potential role for renal TLR2 and TLR4 in the primary mechanism through which the kidney monitors renal injury and initiates and regulates inflammation. Indeed, TLR2 and TLR4 different effects were found in both infection models,36 as in the tissue injury models.37 Because TLR4 can recognize other endogenous ligands than TLR2, they can use an alternative signaling cascade and do not hybridize with other TLR family members as TLR2 does; the functional significance of TLR4 during tissue injury could be different from TLR2.38
The individual role of TLR4 on renal I/R injury was shown by Pulskens et al,39 demonstrating the proinflammatory role of TLR4, as shown by a reduced amount of infiltrating granulocytes and the chemokines in kidneys of TLR4−/− mice compared with wild-type mice. Since an exaggerated inflammatory response could lead to more severe tissue damage, this increased inflammation could explain why TLR4−/− mice showed less tubular injury and a more preserved renal function, compared with wild-type mice upon I/R injury. Epithelial and leukocyte-associated functional TLR4 contribute in a similar proportion to renal dysfunction and injury as assessed by bone marrow chimeric mice. No significant differences were found in renal function and inflammation in both MyD882/2 and TRIF-mutant mice when compared to their wild types, allowing the identification of TLR4 as a cellular sentinel for acute renal damage. These latest data were corroborated by studies from Shigeoka et al19 in TLR2 knockout mice. These animals were better protected against IRI compared to MyD88 knockout mice, indicating that TLR2-dependent, MyD88-independent pathways contribute to kidney injury. In contrast, Wu et al40 in bone marrow chimeric mice studies demonstrated that TLR4 signaling through the MyD88-dependent pathway was required for the full development of kidney IRI, as both TLR4(−/−) and MyD88(−/−) mice were protected against kidney dysfunction, tubular damage, neutrophil and macrophage accumulation, and proinflammatory cytokines release.
Several renal endogenous ligand expressions increased after ischemia reperfusion injury, including biglycan, HMGB1 and hyaluronan,41 providing evidence that one or more of these ligands may be the source of TLR4 activation.
Chen et al42 confirmed the role of TLR4 in ischemic kidney injury using mice harboring spontaneous disabling mutations of the receptor and generated chimeras between TLR4(−/−) and TLR4(+/+) mice. The results of this study demonstrated the necessity of TLR4 in leukocytes, as well as in epithelial and endothelial cells, for the full-blown ischemic response and strongly suggest that the release of HMGB1 from injured epithelia and/or endothelia activates leukocytes to generate proinflammatory cytokines, further exacerbating the injury to ischemic kidneys.41 The sequence of events proposed by Chen et al42 ascribes to the release of HMGB1 from damaged epithelial and endothelial cells the role of a trigger for TLR4-induced activation of leukocytes and macrophages, which subsequently secrete proinflammatory IL-6.42 In a recent study, neutralization of the extracellular HMGB1 released by damaged ischemic renal cells by the administration of blocking antibody to HMGB1 conferred protection against ischemia and reperfusion.43
These recent findings provide an excellent platform for discussing the complexity of danger signaling in the development of AKI.
TLRs and sepsis-induced renal failure
TLRs role in sepsis-induced renal failure is not only limited to the recognition of exogenous PAMPs, but it extends to the recognition of local DAMPs produced during ischemic renal injury and the recognition of extrarenal DAMPs as circulating products of extensive tissue injury in sepsis. The LPS derived from the cell wall of gram-negative bacteria continuously activates TLR4 in a pathogenic septic environment. Loss-of-function mutations in the TLR4 gene in mice are highly resistant to LPS-induced sepsis.44 Bacterial sepsis and acute endotoxemia induce renal tubule malfunction.45 The thick ascending limb may play a role in the innate immune response of the kidney. The TLR4 has been localized in the thick ascending limb.46
TLR4 expression in this segment is increased in response to sepsis and I/R injury and the thick ascending limb has been implicated in mediating inflammatory renal injury during these conditions.46,47 Moreover, the medullary thick ascending limb (MTAL) has been identified as a site of cell damage and tubule dysfunction in response to microbial infection. HCO3− reabsorption is inhibited by LPS in the MTAL from either the basolateral or luminal cell surface through the activation of TLR4.48 Although the underlying signaling mechanisms are different, HCO3− reabsorption through the activation of an extracellular-signal-regulated kinase (ERK)-dependent pathway is decreased in the presence of basolateral LPS.48,49 In addition, basolateral LPS inhibits HCO3− reabsorption in the MTAL through the activation of a TLR4/MyD88/mitogen-activated protein kinase (MEK)/ERK pathway is coupled to the inhibition of sodium–hydrogen exchanger (NHE) 3. NHE3 has been identified as a target of TLR4 signaling in the MTAL.
Thus, bacterial molecules can impair the reabsorptive functions of renal tubules through the inhibition of the exchanger NHE3.50 The ERK pathway links TLR4 to downstream modulation of ion transport proteins, and it has been suggested as a potential target for treatment of sepsis-induced renal tubule dysfunction. In contrast, the lumen LPS inhibition of HCO3− reabsorption was unaffected by ERK inhibitors, but it was eliminated by PI3K inhibitors and Rapamycin, consistent with the activation of a PI3K-mTOR signaling pathway coupled to inhibition of NHE1.51 Thus, the HCO3− reabsorption is inhibited by LPS in the MTAL through the activation of distinct TLR4-mediated signaling pathways in the basolateral and apical membranes. The molecular mechanisms that underlie this membrane-specific TLR4 signal specificity are undefined and have implications for the selective targeting of TLR4-induced inflammatory responses in epithelial cells.48,51
In addition to inhibition by Gram-negative LPS through TLR4, reabsorption of HCO3− by the MTAL is inhibited by gram-positive bacterial molecules through TLR2. The TLR2 is expressed selectively in the basolateral membrane of MTAL cells, in contrast to TLR4, which is expressed in both basolateral and apical membrane domains.49,52 The MTAL HCO3− reabsorption is inhibited by bacterial lipopeptides and gram-positive bacterial cell wall structures (lipoteichoic acid and peptidoglycan) recognized by TLR2. Moreover, the inhibition by gram-positive bacterial components is additive to inhibition by basolateral LPS, due to the TLR2 and TLR4 agonists activating different cell signaling pathways. These findings have important implications for the pathogenesis of kidney dysfunction during polymicrobial sepsis, because they show that gram-negative and gram-positive bacterial molecules can act independently and additively to impair renal tubule function by activating different intracellular signals through different TLRs.52 The separate effects of TLR2 and TLR4 ligands to alter renal tubule function are consistent with the distinct roles of these receptors in bacterial recognition, in which TLR4 is critical for defense against gram-negative infection through the recognition of LPS, and TLR2 plays a predominant role in detection and protection against gram-positive infection.53,54
Recently, Good et al55 reported that TLR2 is required – along with TLR4 – for the response of the MTAL to gram-negative bacterial LPS. Their results showed the effects of basolateral LPS to activate ERK and to inhibit HCO3− absorption are dependent on both TLR4 and TLR2 and provide evidence of an association between TLR4 and TLR2. These findings reveal a novel requirement for TLR2 in LPS-induced TLR4 signaling and suggest that the interaction between TLR4 and TLR2 may play a role in mediating renal tubule dysfunction during sepsis.55
It has been reported that the recognition of LPS by TLR4 is facilitated by the adaptor molecule CD14 and that the binding of MD2 (myeloid differentiation factor 2) in this complex is required for TLR4-LPS signaling.56,57 Soluble CD14 in the urine of patients exhibiting inflammation or sepsis increases sensitivity to LPS. Human proximal tubular epithelial cell death due to sepsis/inflammation showed characteristics of both necrosis and apoptosis, where necrosis was primarily attributable to LPS stimulation, while CD14 was required for induction of renal cell apoptosis.58
In addition, fluorescent microscopy in the rat kidney showed that colocalization of TLR4 and CD14 in renal tubular cells and their expression patterns were markedly affected in response to sepsis.46 LPS injection in pretreated rats with an antagonist of renal beta2-adrenoreceptor (modulator of LPS transport in the kidney), decreased creatinine clearance combined with a significant increase in the expression of TLR4, CD14, and tumor necrosis factor alpha.59 Concurrently, soluble MD2 was found to be increased in the serum and urine of septic patients, and this molecule has been shown to increase TLR4 activation of the receptor in human embryonic kidney epithelial cells in response to LPS.60
Septic AKI is an important clinical syndrome characterized by an abnormal hydroelectrolytic and acid base balance, and it is defined by the simultaneous presence of acute renal failure and sepsis. The understanding of the pathogenesis of septic AKI has been based on the behavior of serum and urinary markers of renal damage and from experimental animal model data. Different cell types and inflammatory mediators are involved in the pathogenesis of organ dysfunction secondary to sepsis.61 Furthermore, the innate immune response is responsible for the initial inflammatory actions in sepsis. As previously discussed, the TLR activation in sepsis-induced renal failure is not only attributed to PAMPs, but it might account for recognition of circulating DAMPs produced subsequent to the extensive tissue injury in sepsis.
Epidemic hemolytic uremic syndrome with diarrhea at presentation (HUS D+) is a major cause of AKI in children.62 Although Shiga toxin (Stx) is the main pathogenic factor and is necessary for epidemic HUS development, the experimental and clinical evidence both suggest that the inflammatory response potentiates Stx toxicity. In fact, both bacterial LPS and polymorphonuclear neutrophils play a key role in the full development of HUS. Therefore, synergism between LPS and Stx2 has been demonstrated as a consequence of the enhancement of Stx2 renal toxicity.63
Recently, we investigated TLR4 surface receptor expression on peripheral blood neutrophils and their ability to modulate inflammatory cytokine release in patients 1 day, 3 days, and 10 days after HUS onset. The isolated leucocytes from the HUS-onset patients exhibited significantly higher mRNA TLR4 expression than the controls. Moreover, TLR4 protein expression on neutrophils determined by flow cytometry was upregulated (Figure 3), driving dependent proinflammatory cytokine, tumor necrosis factor alpha, IL-8 increase, and decreased anti-inflammatory IL-10 release at HUS onset compared with patients with enterohemorrhagic Escherichia coli (EHEC) diarrhea and controls (Figure 4).
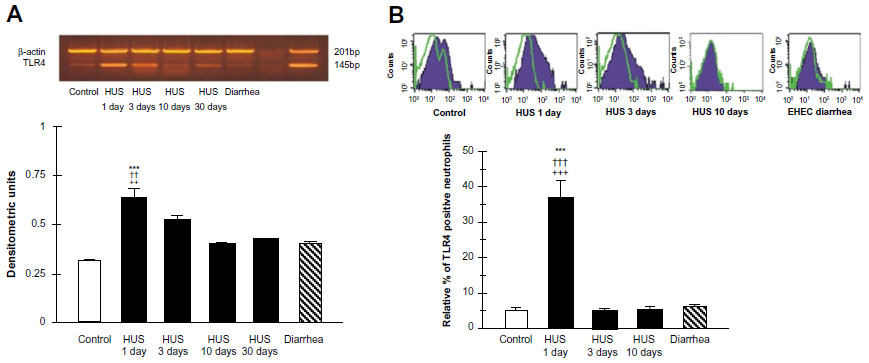 | Figure 3 TLR4 mRNA expression, RT-PCR (A) and protein expression determined by flow cytometry (B) on peripheral blood leukocytes from hemolytic uremic syndrome (HUS) patients, from patients with enterohemorrhagic Escherichia coli (EHEC) diarrhea and healthy controls. |
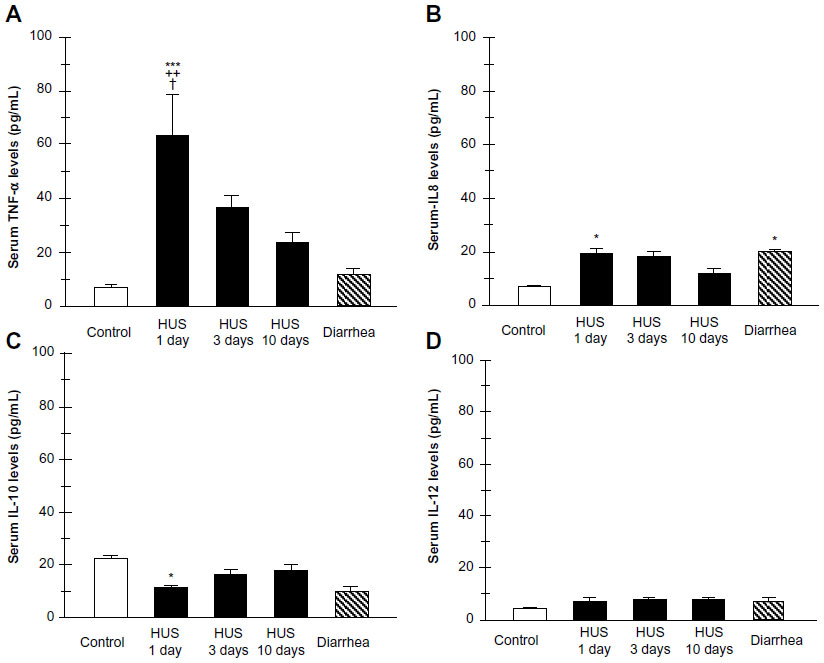 | Figure 4 Plasma cytokine levels from patients within 1, 3, and 10 days of hemolytic uremic syndrome (HUS) onset, from patients with enterohemorrhagic Escherichia coli (EHEC) diarrhea and healthy controls. Supernatants were collected and assayed by enzyme linked immunosorbent assay (ELISA) for tumor necrosis factor alpha (TNF-α) (A), interleukin 8 (IL-8) (B), IL-10 (C), and IL-12 (D) concentrations. |
Conversely, significant reduction of the neutrophil TLR4 receptor expression and lack of cytokine-responsive element activation was shown in patients 3 days and 10 days after the HUS onset. These results suggest that TLR4 expression may be differently regulated on neutrophils. They could be dynamically modulated across the early development of HUS on neutrophils, resulting in the negative regulation preceded by TLR4 overactivation.64
TLRs and cell regeneration
Ischemic injury of the kidney in humans results in tubular cell death and the clinical syndrome of acute tubular necrosis. Tubular cell injury peaks 2–3 days after injury and is followed by a repair phase in which proliferation of surviving tubular cells promotes tubule regeneration. A sterile inflammatory response that contributes to the extent of tubular cell damage is involved in AKI.65 During the early phase of renal injury, leukocyte populations seem to be detrimental, because they promote cell death by apoptosis-releasing reactive oxygen species and activating caspases.66 In sequence, tubular cell death by necrosis is the predominant trigger for this associated inflammatory response, dying cells release intracellular molecules, such as HMGB1, histones, uric acid, or adenosine triphosphate (ATP), that elicit immunostimulatory effects in the extracellular space, referred as DAMPs.28 DAMPs activate a set of pattern recognition receptors, such as TLRs on renal parenchymal cells as well as in interstitial dendritic cells.67 The subsequent innate immune response involves the transcription of numerous proinflammatory cytokines and chemokines, which initiate the influx of various immune cell subsets into the kidney, which contribute to the early amplification of the inflammatory response and AKI by enhancing immune-mediated tubular cell death.68
In the ischemic injury, the inflammatory response is accompanied by the rapid influx of polymorphonuclear leukocytes, lymphocytes, and macrophages into the interstitium. Mononuclear phagocyte/macrophages are one of the major cell types that accumulates around tubules;69 mononuclear inflammatory cells have been demonstrated in the vasa recta of the outer medulla. Macrophages exhibit a range of phenotypes; this phenomenon has been described as macrophage polarization or heterogeneity. It is the effector phenotype of the recruited macrophages rather than their presence alone that determines the extent of renal parenchymal injury as has been suggested in recent studies of other forms of immune-mediated renal injury. It has been proposed that inflammatory cells play a negative role. Interventions that could mitigate the initial inflammatory response by inhibition of the cytokine action, prevention of inflammatory cell homing, or depletion inflammatory cells, seem to decrease the degree of morphologic and functional injury in renal I/R.70
To limit overshooting immunopathology in sterile tissue injuries and allow tissue recovery, a number of counter regulatory mechanisms exist that mostly limit immune activation of intrarenal dendritic cells.71 The phenotypic switching of intrarenal mononuclear phagocytes away from classically activated (proinflammatory) to alternatively activated (anti-inflammatory/proregeneratory) cells is necessary for recovery on AKI. However, the mechanisms by which macrophages are polarized to a trophic phenotype and promote tubular repair after ischemic injury have yet to be fully elucidated.72
Notwithstanding, surviving TECs enter the cell cycle within few hours on injury, a functional tubular recovery does not occur before the resolution of sterile inflammation has occurred and the tubulointerstitial microenvironments become dominated by proregeneratory factors.72 These factors are provided in a paracrine manner by other surviving TECs, intratubular progenitor cells, or bone marrow-derived stem cells.73 The process of repair begins with a marked increase in tubular cell proliferation that peaks on day 3 and slowly declines over the ensuing week. Most dividing cells are tubular cells, with approximately 88% of the bromodeoxyuridine-positive proliferating cells expressing the proximal tubule marker megalin and 1% expressing the thick ascending limb marker Tamm–Horsfall protein. The remaining 10% was unclassifiable. Although dendritic cells and other immune cells play a dominant role in orchestrating the early injury phase of AKI, little is known about the contribution of intrarenal immune cells to the subsequent phase of kidney regeneration.65,73
Recently, Kulkarni et al67 identified yet unknown proregeneratory properties of ILs, a family of leukocyte-derived mediators on TEC regeneration in AKI. They found the IL-22 secretion to be selectively induced by TLR4 agonists released from necrotic tubular cells, which first documents a role of TLR signaling not only for renal immunopathology but also for kidney regeneration in vivo. Furthermore, these data imply that dying TECs involve interstitial dendritic cells to support their regeneration through a specific TLR4–IL-22 pathway. Moreover, the results from Kulkarni et al67 allow to support the idea that the function of TLRs extends beyond host defense against invading pathogens; TLR4 seems to be a cellular sensor for acute renal damage that controls innate immunity and tissue integrity.67 Much more work remains to be done to define more clearly our understanding of how injury and repair are regulated under normal and pathologic conditions.
Disclosure
The authors report no conflicts of interest in this work.
References
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. | |
Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nature Rev Immunol. 2003;3(2):169–176. | |
Medzhitov R. Toll-like receptors and innate immunity. Nature Rev Immunol. 2001;1(2):135–145. | |
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–680. | |
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973–983. | |
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. | |
Medzhitov R, Preston-Hurlburt P, Kopp E, et al. MyD88 is an adaptor protein in the Toll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2(2):253–258. | |
Shirali AC, Goldstein DR. Tracking the toll of kidney disease. J Am Soc Nephrol. 2008;19(8):1444–1450. | |
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–995. | |
Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. | |
Anders HJ, Schlondorff DO. Innate immune receptors and autophagy: implications for autoimmune kidney injury. Kidney Int. 2010;78(1):29–37. | |
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. | |
Baccala R, Gonzalez-Quintial R, Lawson BR, et al. Sensors of the innate immune system: their mode of action. Nat Rev Rheumatol. 2009;5(8):448–456. | |
Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23(11):509–512. | |
Atkinson TJ. Toll-like receptors, transduction-effector pathways, and disease diversity: evidence of an immunobiological paradigm explaining all human illness? Int Rev Immunol. 2008;27(4):255–281. | |
Tesar BM, Goldstein DR. Toll-like receptors and their role in transplantation. Front Biosci. 2007;12:4221–4238. | |
Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5(12):1219–1226. | |
Tsuboi N, Yoshikai Y, Matsuo S, et al. Roles of toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J Immunol. 2002;169(4):2026–2033. | |
Shigeoka AA, Holscher TD, King AJ, et al. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178(10):6252–6258. | |
O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. | |
Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30(1):16–34. | |
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. | |
Ishii KJ, Coban C, Akira S. Manifold mechanisms of Toll-like receptor-ligand recognition. J Clin Immunol. 2005;25(6):511–521. | |
Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420: 885–891. | |
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. | |
Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172(4):2629–2635. | |
Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8(1):11–13. | |
Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol. 2011;22(3):416–425. | |
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. | |
Daha MR, van Kooten C. Is the proximal tubular cell a proinflammatory cell? Nephrol Dial Transplant. 2000;15 Suppl 6:41–43. | |
Leemans JC, Stokman G, Claessen N, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115(10):2894–2903. | |
Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334(22):1448–1460. | |
Kim BS, Lim SW, Li C, et al. Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation. 2005;79(10):1370–1377. | |
Wolfs TG, Buurman WA, van Schadewijk A, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168(3):1286–1293. | |
Mudaliar H, Pollock C, Komala MG, Chadban S, Wu H, Panchapakesan U. The role of Toll-like receptor proteins (TLR) 2 and 4 in mediating inflammation in proximal tubules. Am J Physiol Renal Physiol. 2013;305(2):F143–F145. | |
Arko-Mensah J, Julián E, Singh M, Fernández C. TLR2 but not TLR4 signalling is critically involved in the inhibition of IFN-gamma-induced killing of mycobacteria by murine macrophages. Scand J Immunol. 2007;65(2):148–157. | |
Zhai Y, Shen XD, O’Connell R, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173(12):7115–7119. | |
Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76(3):514–519. | |
Pulskens WP, Teske GJ, Butter LM, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One. 2008;3(10):e3596. | |
Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–2859. | |
Goligorsky MS. TLR4 and HMGB1: partners in crime? Kidney Int. 2011;80(5):450–452. | |
Chen J, Hartono JR, John R, et al. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011;80(5):504–515. | |
Li J, Gong Q, Zhong S. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol Dial Transpl. 2011;26(2):469–478. | |
Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. | |
Chowdhury P, Sacks SH, Sheerin NS. Minireview: functions of the renal tract epithelium in coordinating the innate immune response to infection. Kidney Int. 2004;66(4):1334–1344. | |
El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ, Dagher PC. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am J Physiol Renal Physiol. 2006;290(5):F1034–F1043. | |
El-Achkar TM, Dagher PC. Renal Toll-like receptors: recent advances and implications for disease. Nat Clin Pract Nephrol. 2006;2(10):568–581. | |
Good DW, George T, Watts BA 3rd. Lipopolysaccharide directly alters renal tubule transport through distinct TLR4-dependent pathways in basolateral and apical membranes. Am J Physiol Renal Physiol. 2009;297(4):F866–F874. | |
Good DW, George T, Watts BA 3rd. Nerve growth factor inhibits Na+/H+ exchange and formula absorption through parallel phosphatidylinositol 3-kinase-mTOR and ERK pathways in thick ascending limb. J Biol Chem. 2008;283(39):26602–26611. | |
Watts BA 3rd, George T, Sherwood ER, Good DW. Basolateral LPS inhibits NHE3 and HCO Formula absorption through TLR4/MyD88-dependent ERK activation in medullary thick ascending limb. Am J Physiol Renal Physiol. 2011;301(6):C1296–C1306. | |
Watts BA 3rd, George T, Good DW. Lumen LPS inhibits HCO3(-) absorption in the medullary thick ascending limb through TLR4-PI3K-Akt-mTOR-dependent inhibition of basolateral Na+/H+ exchange. Am J Physiol Renal Physiology. 2013;305(4):F451–F462. | |
Good DW, George T, Watts BA 3rd. Toll-like receptor 2 mediates inhibition of HCO(3)(_) absorption by bacterial lipoprotein in medullary thick ascending limb. Am J Physiol Renal Physiol. 2010;299(3):F536–F544. | |
Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. | |
Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J Dent Res. 2005;84(7):584–595. | |
Good DW, George T, Watts BA 3rd. Toll-like receptor 2 is required for LPS-induced Toll-like receptor 4 signaling and inhibition of ion transport in renal thick ascending limb. J Biol Chem. 2012;287(24):20208–20220. | |
Shimazu R, Akashi S, Ogata H, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. | |
Kitchens RL. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem Immunol. 2000;74:61–82. | |
Bussolati B, David S, Cambi V, Tobias PS, Camussi G. Urinary soluble CD14 mediates human proximal tubular epithelial cell injury induced by LPS. Int J Mol Med. 2002;10(4):441–449. | |
Nakamura A, Niimi R, Yanagawa Y. Renal beta(2)-adrenoceptor modulates the lipopolysaccharide transport system in sepsis-induced acute renal failure. Inflammation. 2009;32(1):12–19. | |
Pugin J, Stern-Voeffray S, Daubeuf B, Matthay MA, Elson G, Dunn-Siegrist I. Soluble MD-2 activity in plasma from patients with severe sepsis and septic shock. Blood. 2004;104(13):4071–4079. | |
Gonçalves GM, Castoldi A, Braga TT, Câmara NO. New roles for innate immune response in acute and chronic kidney injuries. Scand J Immunol. 2011;73(5):428–435. | |
Clayton F, Pysher TJ, Lou R, et al. Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic uremic syndrome. Am J Nephrol. 2005;25(6):536–540. | |
Palermo M, Alves-Rosa F, Rubel C, et al. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin Exp Immunol. 2000;119(1):77–83. | |
Vallés PG, Melechuck S, González A, Manucha W, Bocanegra V, Vallés R. Toll-like receptor 4 expression on circulating leucocytes in hemolytic uremic syndrome. Pediatr Nephrol. 2012;27(3):407–415. | |
Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. | |
Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. | |
Kulkarni OP, Hartter I, Mulay SR, et al. Toll-Like Receptor 4–Induced IL-22 Accelerates Kidney Regeneration. J Am Soc Nephrol. Epub January 23, 2014. | |
Allam R, Scherbaum CR, Darisipudi MN, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23(8):1375–1388. | |
Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66(2):480–485. | |
Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80(9):915–925. | |
Lassen S, Lech M, Römmele C, Mittruecker HW, Mak TW, Anders HJ. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol. 2010;185(3):1976–1983. | |
Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–325. | |
Lech M, Gröbmayr R, Weidenbusch M, Anders HJ. Tissues use resident dendritic cells and macrophages to maintain homeostasis and to regain homeostasis upon tissue injury: the immunoregulatory role of changing tissue environments. Mediators Inflamm. 2012;2012:951390. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.